-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2021; 11(1): 1-7
doi:10.5923/j.chemistry.20211101.01
Received: Nov. 12, 2020; Accepted: Dec. 16, 2020; Published: Jan. 15, 2021

Computational Studies (DFT) and PM3 Theories on Thiophene Oligomers as Corrosion Inhibitors for Iron
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLM. A. Erteeb1, E. E. Ali-Shattle2, S. M. Khalil3, H. A. Berbash3, Z. E. Elshawi3
1Higher Institute of Sciences and Technology - Azizia, Tripoli, Libya
2University of Tripoli, Chemistry Department, Faculty of Science, Tripoli, Libya
3School of Basic Science, Libyan Academy of Graduate Studies, Tripoli, Libya
Correspondence to: E. E. Ali-Shattle, University of Tripoli, Chemistry Department, Faculty of Science, Tripoli, Libya.
| Email: |  |
Copyright © 2021 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Theoretical quantum chemical studies were performed on thiophene oligomers as corrosion inhibitors for iron, by using density functional theory (DFT) at B3LYP/6-31G level (d) and semiemperical methods (PM3) (Parametric Method 3). The correlation between the molecular structure and corrosion inhibitor efficiency of the oligomers were studied. Quantum chemical parameters such as, transferred electronic charge (ΔN), electrophilicity index (ω), chemical softness (σ), absolute electronegativity (χ), electrophilicity index (ω), and chemical hardness (η) of thiophene oligomers were reported. Energy of highest occupied molecular orbitals (HOMO), lowest unoccupied molecular orbitals (LUMO) and energy gap (ΔE = EHOMO - ELUMO) were calculated. The interaction of frontier molecular orbitals with iron orbitals reveal that, charge transfer mechanism may be responsible for the binding or adsorption of these compounds onto the metal surface. It was found that the corrosion inhibitor efficiency of thiophene oligomers increases with an increasing number of thiophene units, in the oligomer structure.
Keywords: Iron, DFT, PM3, Corrosion, Inhibitor, Thiophene Oligomers
Cite this paper: M. A. Erteeb, E. E. Ali-Shattle, S. M. Khalil, H. A. Berbash, Z. E. Elshawi, Computational Studies (DFT) and PM3 Theories on Thiophene Oligomers as Corrosion Inhibitors for Iron, American Journal of Chemistry, Vol. 11 No. 1, 2021, pp. 1-7. doi: 10.5923/j.chemistry.20211101.01.
Article Outline
1. Introduction
- Corrosion has been the subject of scientific study for more than 150 years. It is a naturally occurring phenomenon commonly defined as the deterioration of a material (usually a metal) or its properties because of a reaction with its environment. Like other natural hazards such as earthquakes or severe weather disturbances, corrosion can cause dangerous and expensive damage to everything from pipelines, bridges, and wastewater systems, and even home appliances. Unlike weather-related disasters, however, there are time-proven methods to prevent and control corrosion that can reduce or eliminate its impact on public safety, the economy, and the environment. The science of corrosion prevention and control is highly complex, exacerbated by the fact that corrosion takes many different forms and is affected by numerous outside factors. corrosion professionals must understand the effects of environmental conditions such as soil resistivity, humidity, and exposure to salt water on various types of materials; the type of product to be processed, handled, or transported; required lifetime of the structure or component; proximity to corrosion-causing phenomena such as stray current from real systems, appropriate mitigation methods, and other considerations before determining the specific corrosion problem and specifying an effective solution [1].
1.1. PM3 (Parameterized Model Number 3) Method
- PM3 is another semi-empirical method based on the neglect of differential diatomic overlap integral approximation. The PM3 method uses the same formalism and equations as the Austin Model 1, AM1 method. The only differences are: (a) PM3 uses two Gaussian functions for the core repulsion function, instead of the variable number used by AM1 (which uses between one and four Gaussians per element); (b) the numerical values of the parameters are different. The other differences lie in the philosophy and the methodology used during the parameterization: whereas AM1 takes some of the parameter values from spectroscopic measurements, PM3 treats them as optimized values. The method was developed by Stewart and first reported in 1989 [2].
1.2. Ab Initio and Density Functional Theory (DFT) Methods
- Quantum mechanical methods (ab initio, density functional theory (DFT) and semi empirical PM3) are all based on solving the time independent Schrodinger equation for the electrons of a molecular system as a function of the positions of the nuclei. The term ab initio indicates that the calculation is from first principles and that no empirical data is used.The simplest type of ab initio electronic structure calculation is the Hartree Fock (HF), in which the instantaneous Columbic electron–electron repulsion is not specifically taken into account and only its average effect is included in the calculation. This is a variational procedure, and therefore, the obtained approximate energies, expressed in terms of the system wave function, are always equal to or greater than the exact energy, and approach a limiting value called the Hartree–Fock limit as the size of the basis is increased [3]. Many types of calculations (Moller-Plesset perturbation theory) begin with a Hartree–Fock calculation and subsequently correct for electron–electron repulsion, referred to also as electronic correlation.Density functional theory (DFT) is used to investigate the electronic structure, principally the ground state of many-body systems, in particular atoms, molecules and the condensed phases. The main objective of DFT is to replace the many-body electronic wave function with the electronic density as the basic quantity [4]. Any exchange functional can be combined with any correlation functional in DFT calculations. For example, the notation BLYP/ 6-31G* denotes a density functional calculation done with the Becke 1988 exchange functional and the Lee-Yang-Parr correlation functional, with the orbitals expanded in a 6-31G* basis set [5]. Density Functional Theory (DFT) [6,7] which is an economic and efficient quantum chemistry computing method can provide accurate information about geometrical configuration and electron distribution. DFT is widely applied in the analysis of corrosion inhibition performance and the interaction of corrosion inhibitors and interfaces [8-9]. The B3LYP functional was applied within the context of Gaussian 09 [10], using the 6-31+G (d, p) basis set [11,12]. Optimized structures were verified as minima via the presence of all positive harmonic frequencies. In the aqueous phase calculations, the theoretical model was considered via SMD, using a dielectric constant of 78.5 for water [13a,13b]. Frequency analysis showed there was no imaginary frequency, indicating that the calculated geometry represented a stable minimum on the potential energy surface. The aim of this work is to perform a theoretical calculations on thiophene oligomers (1 to 10) by using DFT and PM3 methods to calculate the quantum chemical parameters such as the chemical hardness (η), the chemical softness (σ), the absolute electronegativity (χ), the electrophilicity index (ω), and the fractions of electrons transferred (ΔN). Also to calculate EHOMO, ELUMO, ΔE = (ELUMO-EHOMO) to find a correlation between these parameters and the efficiency of thiophene oligomers (1 to 10) as corrosion inhibitors for iron metal.
2. Results and Discussion
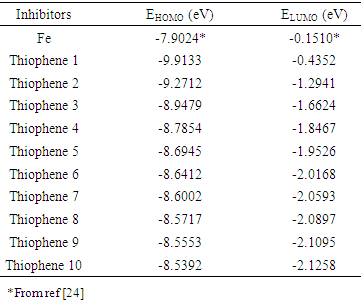
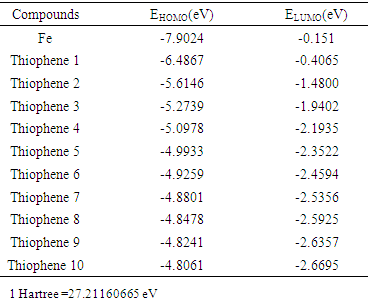
- The thiophene Oligomers under investigation and its structures are shown in Scheme. 1, and the energy of the frontier molecular orbitals, (EHOMO and ELUMO), the energy gap (ΔE), the chemical softness (σ), the chemical hardness (η), and the fraction of the electron transferred (ΔN), and elecrophilicity index (ω), were calculated according to molecular orbital theory [14]. The EHOMO and ELUMO of the inhibitor are related to ionization potential (I) and electron affinity (A) respectively, with reversed sign.
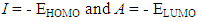
 | Scheme 1. Optimized molecular structures of thiophene oligomers are given in figures (1-10) |
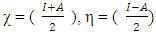 The global softness (σ) is the inverse of the chemical hardness [16].
The global softness (σ) is the inverse of the chemical hardness [16]. Electronegativity, hardness and softness have been proved to be very useful quantities in the chemical reactivity theory. When two systems, metal and inhibitor, are brought in contact together, electrons will flow from lower (χ) inhibitor to higher (χ) metal, until the chemical potentials become an equal. The fraction of the transferred electronic charge (ΔN) from the inhibitor molecule to the metallic atom was calculated according to Pearson formula [17]. For a reaction of two systems with different electronegativities (as a metallic surface and an inhibitor molecule). The following mechanism will take place: the electrons will flow from the molecule of lower electronegativity value toward that of higher value, until the chemical potentials are the same. The global electrophilicity index was introduced by Parr et al.:
Electronegativity, hardness and softness have been proved to be very useful quantities in the chemical reactivity theory. When two systems, metal and inhibitor, are brought in contact together, electrons will flow from lower (χ) inhibitor to higher (χ) metal, until the chemical potentials become an equal. The fraction of the transferred electronic charge (ΔN) from the inhibitor molecule to the metallic atom was calculated according to Pearson formula [17]. For a reaction of two systems with different electronegativities (as a metallic surface and an inhibitor molecule). The following mechanism will take place: the electrons will flow from the molecule of lower electronegativity value toward that of higher value, until the chemical potentials are the same. The global electrophilicity index was introduced by Parr et al.: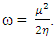 Where μ represent the chemical potential and equal to the negative value of electronegativity χ [20]. According to the definition, this index measures the propensity of chemical species to accept electrons.The following formula was used to calculate ΔN. [18]
Where μ represent the chemical potential and equal to the negative value of electronegativity χ [20]. According to the definition, this index measures the propensity of chemical species to accept electrons.The following formula was used to calculate ΔN. [18] 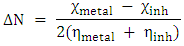 Where χmetal and χinh denote the absolute electronegativity of metal and inhibitor molecule respectively, ηmetal and ηinh denote the absolute hardness of metal and the inhibitor molecule respectively. The difference in electronegativity drives the electron transfer, and the sum of the hardness parameters acts as a resistance [1].
Where χmetal and χinh denote the absolute electronegativity of metal and inhibitor molecule respectively, ηmetal and ηinh denote the absolute hardness of metal and the inhibitor molecule respectively. The difference in electronegativity drives the electron transfer, and the sum of the hardness parameters acts as a resistance [1].2.1. PM3 Calculations
|
|
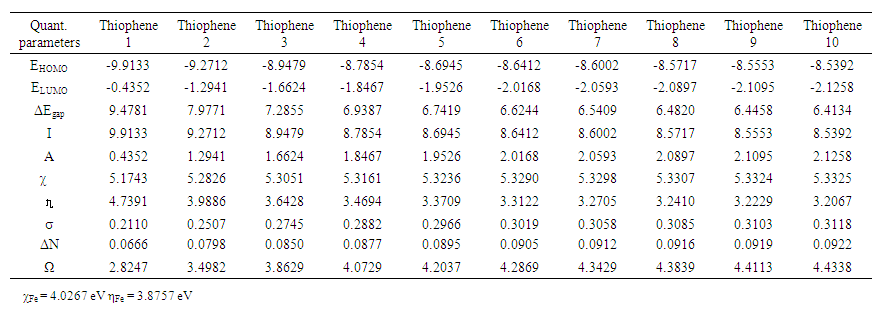 | Table 3. Calculated quantum chemical parameters for the Thiophene oligomers inhibitors for iron |
2.2. Density Functional Theory (DFT) Calculations
- Calculated (HOMO –LUMO) energies of the thiophene oligomer inhibitors by DFT method shown in. Table 4.
|
|
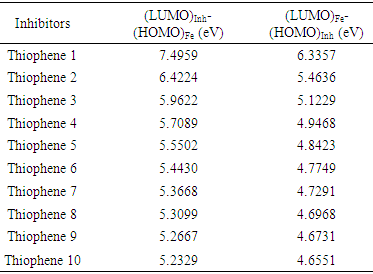
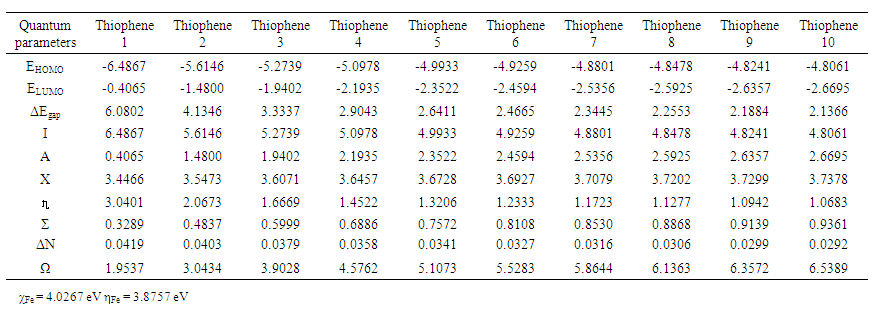 | Table 6. Calculated quantum chemical parameters for (1- 10) thiophene oligomers |
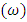 increases as the number of pyrrole molecules increases, suggesting that an increase in the ability of the thiophene oligomers to accept electrons from iron will be increased [39].
increases as the number of pyrrole molecules increases, suggesting that an increase in the ability of the thiophene oligomers to accept electrons from iron will be increased [39].3. Conclusions
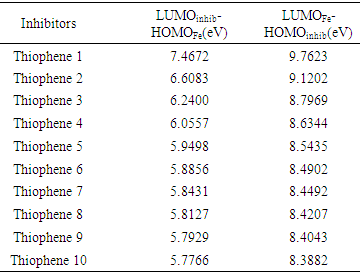
- The present theoretical study indicates the following facts or observations:- It was found that the interactions of the thiophene oligomers with iron have certain ionic character by both PM3 and DFT methods, because the values of LUMOinh- HOMOFe gap fall between 5 to7 eV, by PM3 calculations, and between 4 to 6 eV, DFT calculations. Strong covalent bond can be expected only if LUMOinh- HOMOFe gap is approximately zero eV.- DFT and PM3 calculations have shown a better interaction by increasing the number of the thiophene unit in the oligomers. - Thiophene Oligomer will therefore be a good and effective inhibitor. This agrees with the recently published experimental results [40]. Also, this study, displays a good correlation between corrosion inhibitor efficiency and number of thiophene unit and confirms the reliability of the theoretical methods to study the corrosion inhibitors for metal surfaces.