-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2019; 9(5): 150-158
doi:10.5923/j.chemistry.20190905.03

Some Antibacterial and Antifungal Compounds from Root Bark of Rhus natalensis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLPeter W. Njoroge, Sylvia A. Opiyo
Department of Physical and Biological Sciences, Murang’a University, Murang’a, Kenya
Correspondence to: Sylvia A. Opiyo, Department of Physical and Biological Sciences, Murang’a University, Murang’a, Kenya.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Phytochemical studies on Rhus natalensis root bark collected from Iten, Kenya led to isolation and characterization of five compounds namely epicatechin (1), 3β-sitosterol (2), 3β-sitosterol glucoside (3), stigmasterol (4) and lupeol (5). The structures of the isolated compounds were established using spectroscopic techniques as well as by physical methods. The compounds were tested for bacterial and antifungal activities against Staphylococcus aureus (ATCC no. 25923), Bacillus subtilis (local isolate), Escherichia coli (ATCC no. 25922), Pseudomonas aeruginosa (ATCC no. 10622), Candida albicans, Penicillium notatum and Aspergillus Niger. All the compounds showed antibacterial and antifungal activities against the test pathogens. Compounds 1, 3 and 5, exhibited moderate activity while compounds 2 and 3 showed low activity. These results further confirm the efficacy of extracts of Rhus species in treating bacterial and fungal infections.
Keywords: Rhus species, Isolation and characterization, NMR, Antibacterial, Antifungal, Inhibition zone
Cite this paper: Peter W. Njoroge, Sylvia A. Opiyo, Some Antibacterial and Antifungal Compounds from Root Bark of Rhus natalensis, American Journal of Chemistry, Vol. 9 No. 5, 2019, pp. 150-158. doi: 10.5923/j.chemistry.20190905.03.
Article Outline
1. Introduction
- Bacteria and fungi are frequent causes of serious opportunistic infections [1]. This is more so with a wide range of hosts with weakened immunity such as cancer patients, premature infants and human immunodeficiency virus (HIV) victims [2]. Many opportunistic bacteria and fungi are resistant to the antibiotics currently available in the market [3]. The increase in drug-resistance cannot be matched by parallel expansion in the anti-infective agents used to treat infections [4]. In addition, side effects and cost of antibiotics limit their usefulness in disease management.About 80% of the world’s inhabitants still rely on traditional medicines based on herbal plants for their primary healthcare [5,6]. Previous studies have shown that in plants there are bioactive compounds that have gained increasing interest as potential therapeutic agents [7-9]. Anti-infective agents from plant origin are preferred since they have no side effects, have better patient tolerance and are relatively inexpensive [10]. The genus Rhus (sumac) which belongs to the family Anacardiaceae, consists of approximately 250 species which occur mainly in the tropics, subtropics and temperate areas, especially in North America and Africa [11]. In traditional medicine, extracts of Rhus species are used to manage several ailments including influenza, wounds, diarrhea, abdominal pain, indigestion [12], diabetes, malaria, rheumatism [13], toothaches, swollen legs [14], dog bite, peptic ulcer, kidney stones, skin eruptions, bruises and boils [15]. Root extracts of R. natalensis are used to manage venereal diseases, heartburn, cold, cough, diarrhea [16], hernia and stomach ache [17], influenza, abdominal pain and gonorrhea [12]. The leaves are used for treatment of syphilis [18], cough and colds [12].Previous studied showed that the species posses a wide range of bioactivities including antimicrobial [19-21], antiviral [21], antiinflammation [23], anticancer [24,25], antiplasmodial [26,27] and antioxidant [20,23]. Phytochemical studies revealed that Rhus species are rich in flavonoids [20,23,25,26], biflavonoids [27], anthocyanins and quinones [26], triterpenes [28] sterols and urushiols [20]. We report the antibacterial and antifungal activities of compounds from R. natalensis.
2. Materials and Methods
2.1. General Experimental Procedure
- Melting points were determined on a Gallenkamp (Sanyo, West Sussex, UK) melting point apparatus and are uncorrected. The UV spectra were run on PyeUnicam SP8-150 UV–vis spectrophotometer (Cambridge, UK) using acetonitrile as a solvent. IR data were recorded on a Perkin-Elmer FTIR 593 series spectrophotometer (Tokyo, Japan) on KBr pellets. The 1H and 13C NMR data were recorded in CD3OD on a Varian Gemini (Illinois, USA) operating at 400 MHz, and 100 MHz, respectively. The mass spectroscopy data were obtained on a Varian MAT 8200A instrument (Bremen, Germany).
2.2. Plant Material
- The root bark of R. natalensis was collected from Kapkonga Iten, 40 km north east of Eldoret Town in Kenya. The plant materials were identified at the National Museums of Kenya where the voucher specimen, reference numbers PKT/R1, was deposited. The plant materials were dried under the shade at room temperature for one month and then ground into coarse using a mill (Christy and Norris Ltd., Chelmsford, England).
2.3. Solvent Extraction
- Powdered root bark of Rhus natalensis (1.6 kg) were extracted sequentially with hexane, CH2Cl2, EtOAc and MeOH at room temperature for five days with occasional shaking. The macerates were filtered and concentrated at reduced pressure at 45°C using rotary evaporator to give 12.0, 11.5, 21.5 and 400.0 g of hexane, dichloromethane, ethyl acetate and methanol extracts, respectively. The extracts were stored at 4°C in brown glass bottles.
2.4. Fractionation of Hexane Extract
- Hexane extract (11 g) was dissolved in minimum amount of hexane and adsorbed into silica gel and then evaporation to dryness. The resulting material was loaded on silica gel column (3 cm 60 cm, SiO2 250 g, pressure = 1 bar) and eluted with hexane, hexane-DCM (4:1 and 1:1) and finally with DCM. A total of 153 fractions (each 20 ml) were sampled and their homogeneity was monitored by TLC. The fractions that showed similar TLC profiles were combined to give three pools (I–III). Pool I (fractions 20-62, 211 mg), a yellowish-brown oil, did not give any major spot and was discarded. Pool II (fractions 65-89, 156 mg) which resulted from elution with hexane-DCM 4:1, was subjected to further column chromatography over silica gel, eluting with hexane: DCM, 2:1 to yield compound 2 (24 mg). Pool III (fractions 90-128, 125 mg) which resulted from elution with hexane - DCM 1:1 solvent system was subjected to further column chromatography eluting with hexane-DCM 4:1, 3:2 and 1:1 solvent systems to give 46 fractions (15 ml each). The fractions were combined into two pools (IIIa and IIIb) depending on their TLC profiles. Pool IIIb (hexane-DCM 1:1 eluates, 85.4 mg) was subjected to PTLC fractionation using hexane-DCM 3:1 solvent system to yield 15 mg of compound 4.DCM extract (10.8 g) was fractionated over silica gel column using hexane, gradient hexane-DCM mixture and DCM to give 200 fractions (20 ml each). The fractions were combined into five pools (I-V) depending on their TLC profiles. Pool II (fractions 42-78, 120.0 mg) which resulted from elution with hexane was subjected to further column chromatography eluting with hexane-DCM 2:1 solvent mixture to yield 41 mg of compound 5. Pool III (fractions 80-146, 214 mg) which resulted from elution with hexane- DCM 2:3 solvent system was subjected to further column chromatography and eluted with hexane-DCM 2:3, 1:1, 3:2 and 1:9 solvent systems to give 67 fractions (15 ml each). The fractions were combined into four pools (IIIa-IIId) depending on their TLC profiles. Pool IIIc (fractions 30-42 91.7 mg) which resulted from elution with hexane-DCM 2:3 solvent system crystallized in MeOH to yield 16.9 mg of compound 3. Ethyl acetate extract (20.5 g) was fractionated over silica gel using DCM, DCM containg increaing amounts of EtOAc and finally with EtOAc to give a total of 100 fractions (50 ml each). The fractions were combined into four pools (I-IV) based on their TLC profiles. Pool II (fractions 15-38, 3.0 g) was further fractionated over Sephadex using DC and DCM-MeOH (9:1, 4:1, 3:2 & 1:1) solvent systems to give 32 fractions which were combined into three pools (IIa-IIc) after monitoring their compositions by TLC. Pool IIc (fractions 25-32, 254 mg) was subjected to PTLC fractionation using DCM-acetone 1:1 solvent system to yield 44 mg of compound 1.
2.5. Physical and Spectroscopic Data
- Compound 1: Yellow crystals, mp 115-117°C, Rf 0.68 (acetone: DCM 2:3); 1H-NMR (CD3OD, 400 MHz) δ ppm: 4.59 (1H, d, J = 7.5 Hz), 4.01(1H, m), 2.7 (1H, dd, J = 16.0, 7.0 Hz), 2.53 (1H, dd, J = 16.0, 8.0 Hz), 5.89 (1H, d, J = 2.5 Hz), 5.96 (1H, d, J = 2.5 Hz), 6.87 (1H, d, J = 1.5 Hz), 6.76 (1H, dd, J = 9.5, 1.5 Hz), 6.74 (1H, d, J = 0.5 Hz); 13C NMR (CD3OD, 100 MHZ) δ ppm (Table 1). EIMS (rel. int) m/z: 290, 272, 152 and 139.Compound 2: White crystalline solid, mp 128-130°C, Rf 0.56 (100% DCM). IR νmax (KBr, cm-1) 3422, 2920, 1640, 1463, 1380, 1048, 1021, 723. 1H NMR (CD3OD, 400 MHz) δ ppm: 5.37 (1H, d, J = 7.2 Hz), 3.53 (m), 1.03 (3H, s), 0.94 (3H, d, J = 8.4 Hz), 0.86 (9H, m), 0.70 (3H, s); 13C NMR (CD3OD, 100 MHZ) δ ppm (Table 2). EIMS (rel. int) m/z: 414, 396, 303, 329 and 213.Compound 3: White needle-like crystals, mp 156-160°C, Rf 0.65 (DCM: MeOH 3:1). 1H-NMR (CD3OD, 400 MHz) δ ppm: 5.37 (1H, d, J = 7.2 Hz), 3.53 (m), 1.03 (3H, s), 0.94 (3H, d, J = 8.4 Hz), 0.86 (9H, m), 0.70 (3H, s), 5.2, 3.8, 3.2; 13C NMR (CD3OD, 100 MHZ) δ ppm (Table 2).Compound 4: White feather-like crystals, mp 168-170°C, Rf 0.66 (100% DCM). 1H NMR (CD3OD, 400 MHz) δ ppm: 5.15 (1H), 5.09 (1H, d, J = 1.5, 8.5 Hz), 5.35 (1H, d, J = 3.15 HZ), 3.51(1H, m) 1.01, 0.91, 0.85, 0.81, 0.77, 0.68; 13C NMR (CD3OD, 100 MHZ) δ ppm (Table 2). EIMS (rel. int): m/z 412, 394, 369, 351, 300, 271 and 255. Compound 5: White needle-like crystals, mp 126-128°C, Rf 0.57 (n-hexane: DCM 1:1). 1H NMR (CD3OD, 400 MHz) δ ppm: 4.6, 4.5, 3.15, 2.3, 1.66, 1.01, 0.95, 0.92, 0.81, 0.77 and 0.74 ppm; 13C-NMR (CD3OD, 100 MHZ) δ ppm (Table 2). EIMS (rel. int): m/z 426, 411, 385, 355, 220, 218, 207, 189.
2.6. Antibacterial Activity Test
- Antibacterial test was done using paper disc diffusion method [29]. Staphylococcus aureus (ATCC no. 25923), Bacillus subtilis (local isolate), Escherichia coli (ATCC no. 25922) and Pseudomonas aeruginosa (ATCC no. 10622) used were obtained from the Department of Botany, Kenyatta University in Kenya. Sterile nutrient agar (28 g/l in distilled water, 15 ml) was dispensed in 90 mm diameter sterile petri dishes to yield a uniform depth of 4 mm. Bacteria from stock cultures were spread on the nutrient agar surface using sterile wire loop and incubated at 37°C for 24 hrs. One loop-full of the bacterial strain from the culture was added to sterile nutrient broth medium and incubated at 37°C in a rotary shaker. After 24 hrs, the broth bacteria culture (0.1 ml) was pipetted into nutrient agar media in petri dishes and spread evenly. 100 µl of the isolated compounds at a concentration of 1 mg/ml in DMSO were dispensed on 6 mm sterile paper disc [29]. The discs were oven died at 50°C for 1 hr and then firmly placed on the inoculated petri dishes using sterile forceps. The petri-dishes and their contents were then incubated at 37°C in inverted position. The zones of inhibition were measured after 24 hrs [20,30].
2.7. Anti-fungal Activity Test
- Agar-well diffusion method was used as described by Opiyo et al [31]. Three fungal species, Candida albicans, Penicillium notatum and Aspergillus Niger were obtained from the Department of Botany, Kenyatta University in Kenya. 15 ml of sterile potato dextrose agar prepared by dissolving 39 g in 1L of water was dispensed in petri dishes under sterile conditions and left to solidify. Pure cultures of test fungi were introduced in the petri dishes from stock cultures and incubated at 30°C for 7 days to produce a good crop of spores. The fungal inoculums were prepared by harvesting the spores from the crop of spores using a spores-harvesting needle and transferred into a sterile tube containing sterile distilled water. The spore suspension (0.5 ml) was transferred to PDA surface in petri-dishes using a pipette and the petri-dishes were tilted several times to spread the inoculum. After 10 minutes four wells were cut out from the inoculated PDA medium using a sterile cork-borer (6 mm). 100 μl of suspension of the isolated compounds at concentration of 1 mg/ml in DMSO were pipetted into the wells and then incubated at 30°C for 24 hours. Diameters of zones of inhibition were measured as described by [31].
3. Results and Discussion
3.1. Characterization of Compounds
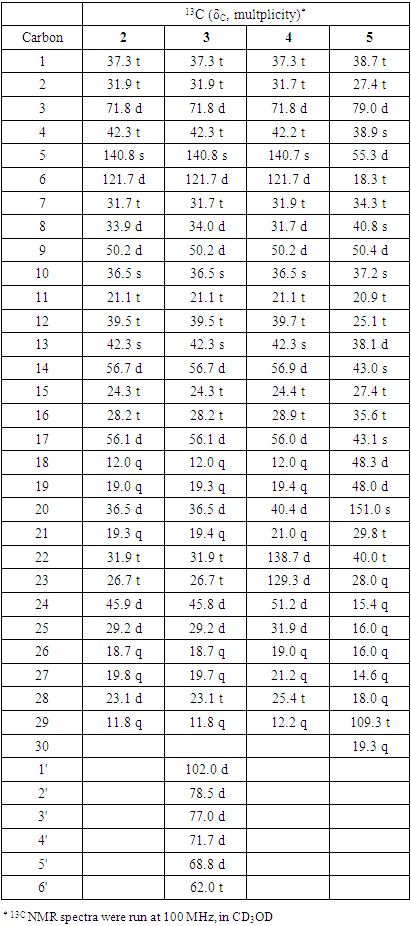
- Chromatographic fractionation of root bark extracts of R. natalensis lead to isolation of five compounds (Figure 1). The structures of the compounds were determined using spectroscopic as well as physical methods. Compound 1 was isolated as yellow crystals having Rf value of 0.68 (DCM-acetone 3:2). On spraying with anisaldehyde the spot turned red and then yellow suggesting the compound to be a flavonoid. 1H-NMR data (Table 1 and Figure 2) showed aromatic peaks at δ 6.87 (1H, d, J = 1.5 Hz), 6.76 (1H, dd, J = 9.5, 1.5 Hz) and 6.74 (1H, d, J = 0.5 Hz) representing protons of a trisubstituted benzene ring, and δ 5.96 (1H, d, J = 2.5 Hz) and δ 5.89 (1H, d, J = 2.5 Hz) representing protons of a tetra-substituted benzene ring [32,33].
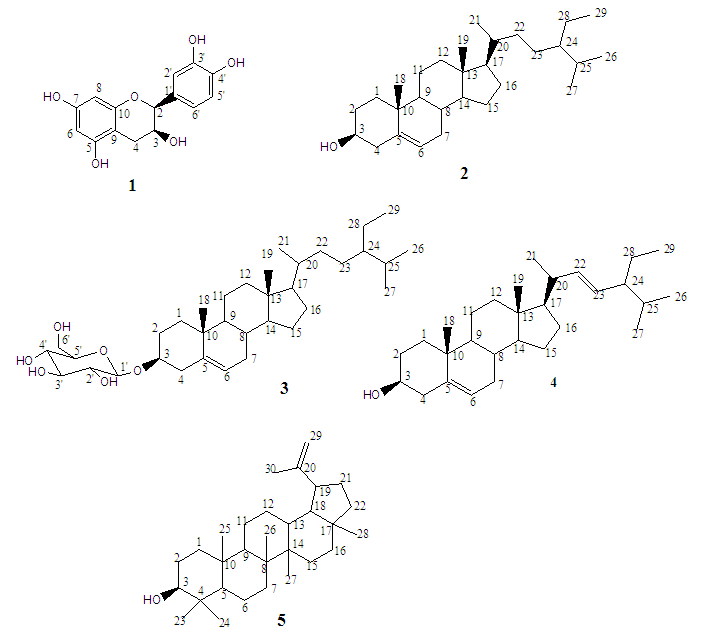 | Figure 1. Structures of the isolated compounds |
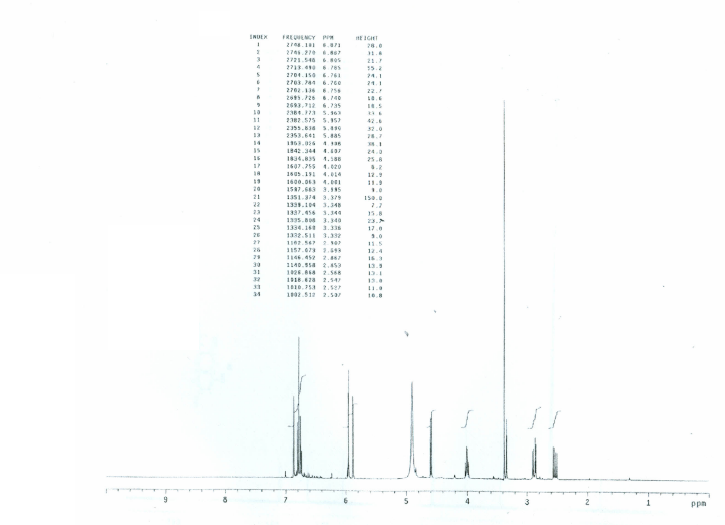 | Figure 2. 1H-NMR spectrum of compound 1 (CD3OD, 400 MHz) |
|
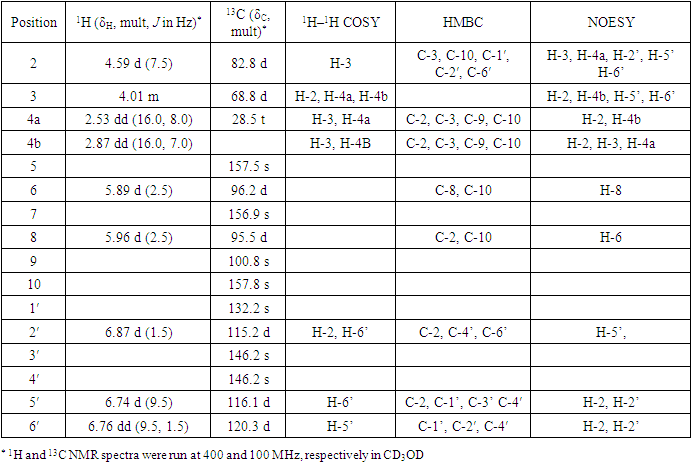
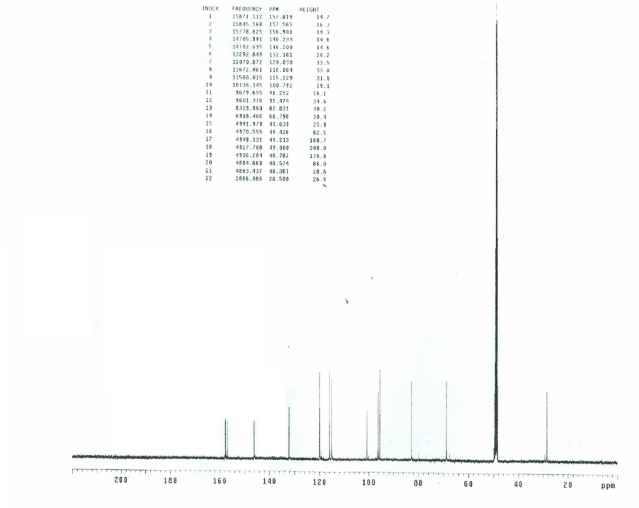 | Figure 3. 13C-NMR spectrum of compound 1 (CD3OD, 100 MHz) |
|
3.2. Antibacterial Activity of Isolated Compounds
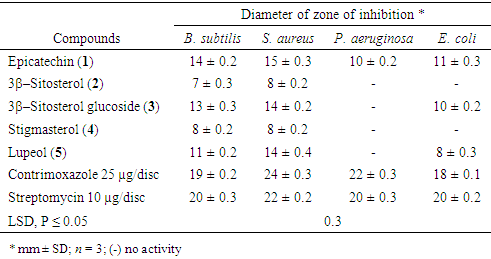
- Isolated compounds were tested for antibacterial activity against P. aeruginosa, B. subtilis, S. aureus and E. coli using paper disc diffusion method. All isolated compounds were active against all the gram-positive bacteria tested (Table 3). Compounds 1, 3 and 5, exhibited moderate activity while 2 and 3 showed low activity. Bacillus subtilis and S. aureus were most susceptible (P ≤ 0.05) to compound 1 with zones of inhibitions of 14 ± 0.2 and 15 ± 0.3 mm respectively. Stigmasterol (4) showed equal antibacterial activity against the two gram-positive bacteria. S. aureus was more susceptible to compound 2 than B. subtilis. Only compounds 1, 3 and 5 were active against the gram-negative bacteria. Escherichia coli was most susceptible (P ≤ 0.05) to epicatechin (1) while P. aeruginosa was only susceptible to epicatechin (1). All isolated compound showed lower antibacterial activity compared to standard drugs. These results were consistent with previous studies which reported antibacterial activities of 3β–sitosterol (2), stigmasterol (4) [36], 3β-sitosterol glucoside (3) [45] lupeol (5) and epicatechin (1) [46].
|
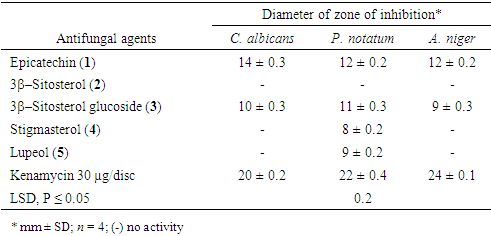
3.3. Antifungal Activity of Compounds
- Isolated compounds were tested for antifungal activity against C. albicans, P. notatum and A. niger (Table 4). Epicatechin (1) recorded the highest activity against all the testes fungi having inhibition zones of 14 ± 0.3, 12 ± 0.2 and 12 ± 0.2 mm against C. albicans, P. notatum and A. niger respectively. Lupeol (5) and stigmasterol (4) were only active against P. notatum with inhibition zones of 9 ± 0.2 and 8 ± 0.2 mm, respectively. β-Sitosterol (2) showed no activity against the test fungi. Antifungal activity of β-sitosterol (2) [36,47], stigmasterol (4) [36], lupeol (5) [48] and β-sitosterol glucoside (3) [45] were earlier reported.
|