-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2015; 5(3): 86-89
doi:10.5923/j.chemistry.20150503.03
On the Regiochemistry in the Heyns Rearrangement
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFrancisco Sánchez-Viesca, Reina Gómez
Department of Organic Chemistry, Faculty of Chemistry, National Autonomous University of Mexico, Mexico City, México
Correspondence to: Francisco Sánchez-Viesca, Department of Organic Chemistry, Faculty of Chemistry, National Autonomous University of Mexico, Mexico City, México.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
The regiochemistry in the Heyns rearrangement has not been explained. Therefore, we studied fructosazone formation, since it involves a Heyns rearrangement. We provide a novel reaction mechanism in order to explain the observed regioselectivity. The proposed reaction mechanism is in accordance with experimental facts and with well known reactivities. Notwithstanding the several studies available on osazone formation, there was a major theoretical gap, a missing link between fact and theory.
Keywords: Fructosazone, Heyns rearrangement, Reaction mechanisms, Reactive intermediates, Regioselectivity
Cite this paper: Francisco Sánchez-Viesca, Reina Gómez, On the Regiochemistry in the Heyns Rearrangement, American Journal of Chemistry, Vol. 5 No. 3, 2015, pp. 86-89. doi: 10.5923/j.chemistry.20150503.03.
1. Introduction
- There are several studies on the mechanism of osazone formation, and are commented in the next section.In the case of glucosazone formation there is no reaction alternative in order to form a vicinal bisphenyl-hydrazone. The osazone is formed at C-1 and C-2. In fructose, the carbonyl group at C-2 could give a vicinal bisphenyl-hydrazone at C-1, C-2 or at C-2, C-3. However, there is further reaction only at C-1, giving the same osazone that is obtained with glucose. This regioselectivity has not been explained previously. In this communication, we provide a reaction mechanism in order to explain the formation of the obtained product in this variant of the Heyns rearrangement.
2. Experimental Facts and Theories
- In glucosazone formation, there is an oxidation of the hydroxymethylene group adjacent to the first obtained phenylhydrazone. Then, the second phenylhydrazone can be formed. It was considered that the oxidation of the involved secondary alcohol was performed by a phenylhydrazine molecule, since three molecules of this reagent are required. This point of view was discarded because phenylhydrazine has not oxidizing properties [1].In order to explain glucosazone formation, two alternative routes, A and B, were proposed by F. Weygand [2, 3] and have been reviewed [4, 5].The oxidation step proposed by Weygand, which is common to both routes, is of interest to us because it does not explain the regioselectivity in fructosazone formation. He considers that in glucosazone formation there is an internal oxido-reduction that can be explained invoking an Amadori rearrangement, i.e., the isomerization of the N-glycoside of an aldose (glycosylamine) to the corresponding 1-amino-1-deoxy-ketose [6-8], Figure 1.
 | Figure 1. Amadori rearrangement |
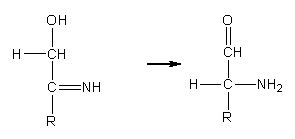 | Figure 2. Heyns rearrangement |
3. Discussion
- Fructose gives an osazone at C-1, C-2. But, being the carbonyl group at C-2, why an osazone at C-2, C-3 is not formed? This regioselectivity not only has not been explained, it has been overlooked.We provide a reaction mechanism that explains the observed regiochemistry. Our proposal is based in very well known reactivities and in related reactions, and also in molecular modelling, as we will see.It is established that the reaction of an α-hydroxycarbonyl compound (an α-ketol) and an arylhydrazine gives the arylhydrazone. The problem is why, in fructosazone formation (a variant of the Heyns rearrangement), the reaction proceeds to C-1 and not to C-3. At this step, the only existing difference is that at C-1 there is a primary alcohol, and at C-3 there is a secondary one. Their respective reactivities are: in acidic medium, the secondary alcohol can be dehydrated more easily than a primary one; but, in the presence of a base, a primary alcohol is more reactive than a secondary one [13, 14]. Thus, the greater acidity of the primary alcohol in the hydroxymethyl group (C-1), must be the determinant factor in the next step. Both hydroxy groups, at C-1 and at C-3, can form a hydrogen bond with the arylhydrazone β-nitrogen. However, from these two possibilities, the hydrogen bond with the primary alcohol at C-1 is preferred because this OH is a better hydrogen donor due to its greater acidity, Figure 3.
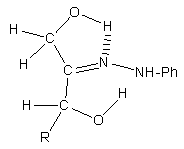 | Figure 3. Preferred hydrogen bond formation in fructose phenylhydrazone |
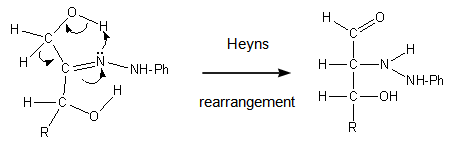 | Figure 4. Concerted oxido-reduction step in fructose phenylhydrazone |
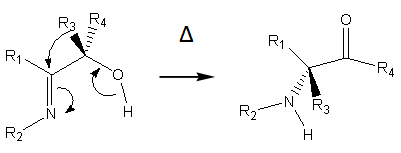 | Figure 5. Thermal rearrangement of α-hydroxy imines |
 | Figure 6. 3D Molecular models of fructose phenylhydrazone, without and with different hydrogen bonds |
 | Figure 7. Fructosazone formation, following steps |
4. Conclusions
- ● A further reaction in fructose phenylhydrazone is an oxido-reduction at C-1, C-2.● This reaction has been explained previously by simple prototropy.● The above point of view does not account for the observed regiochemistry, i.e., why the reaction does not proceed to C-3.● We provide a reaction mechanism that explains the reaction selectivity.● We found that the factor that determines the regiochemistry in this reaction is the different type of alcohol: primary at C-1 and secondary at C-3.● This is due to their different reactivity towards a base.● The β-nitrogen in fructose phenylhydrazone is the Lewis base capable to form a hydrogen bond with a hydroxy group at a vicinal carbon atom.● The preferred hydrogen bond is formed with the OH at C-1 due to the major acidity of this OH (better hydrogen donor).● The oxido-reduction reaction that occurs at C-1, C-2 can then be explained by a 1,4-hydrogen transfer via a cyclic, concerted five-member reaction mechanism (internal catalysis).● Thus, the invoked factors explain completely the selectivity observed in this oxido-reduction step and therefore the regiochemistry of fructosazone. We have cleared a process that has eluded explanation for too many years.