-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2015; 5(2): 49-54
doi:10.5923/j.chemistry.20150502.01
Nucleophilic Substitution at Thiophosphoryl Center (P=S)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLShuchismita Dey
Department of Textile Engineering, Southeast University, Tejgaon, Dhaka, Bangladesh
Correspondence to: Shuchismita Dey, Department of Textile Engineering, Southeast University, Tejgaon, Dhaka, Bangladesh.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Two main types of displacement processes are well known at neutralthiophosphoryl group transfer reactions: concerted involving displacementat phosphorus through a single pentacoordinate transition state (TS) and stepwise mechanism involving a trigonal bipyramidal pentacoordinate (TBP-5C) intermediate. In some cases mechanistic change from concerted to stepwise or vice versahas been reported. This paper includes a review on mechanism of nucleophilic substitution at phosphorus center with P=S substrates. For example the reactions of aryl phenyl chlorothiophospahtes with anilines were reported as concerted whereas the reactions between thiophosphinyl and thiophosphonyl chlorides and R2NH proceed through stepwise mechanism forming thiophosphene intermediates. In case of reactions of O-aryl methylphosphono chloridothioates with anilines the mechanism reported as change from concerted to stepwise. These conclusions were done based on Hammett and Brönsted constants, Cross-interaction Constants and Kinetic Isotope Effects. The papers reported from 1994-2014 were reviewed in this article.
Keywords: Thiophosphoryl transfer reaction, Aminolysis, Pyridinolysis, Concerted, Stepwise
Cite this paper: Shuchismita Dey, Nucleophilic Substitution at Thiophosphoryl Center (P=S), American Journal of Chemistry, Vol. 5 No. 2, 2015, pp. 49-54. doi: 10.5923/j.chemistry.20150502.01.
Article Outline
1. Introduction
- Organothiophosphorous compounds are useful in agricultural, pharmaceuticals and Textile chemicals [1a, b]. Organothiophosphorous compounds are used as pesticides in crop production in agriculture. These substances are also used as flame retardant agent in Textiles. Considering its wide uses in different sectors the displacement reactions at P=S center has become significant in the field of reaction mechanism. The chemistry of organothiophosphate compounds is very important because of greater interest. A significant number of works have been reported on thiophosphoryl (P=S) transfer reactions. Because predicting the effects of replacing the oxygen atom in the phosphoryl group (P=O) with sulfur is necessary. Two main types of displacement processes are well known at neutralthiophosphoryl group transfer reactions: concerted involving displacementat the phosphorus through a single pentacoordinate transitionstate (TS) and stepwise mechanism involving a trigonal bipyramidal pentacoordinate (TBP-5C) intermediate (Scheme 1). In some cases mechanistic change from concerted to stepwise or vice versa has been reported. This paper includes a review on mechanism of nucleophilic substitution at phosphorus center with P=S substrates. Most recent and updated papers have been reviewed in this article to get a clear idea of researchers in this field.
2. Results and Discussion
2.1. Concerted Mechanism
- Dey et al. [1c] reported the mechanism of thiophosphoryl transfer, as well as to compare the reactivity when the oxygen atom in the phosphoryl group is replaced with sulfur, in the aminolyses of aryl phenyl chlorothiophosphates and 4-chlorophenyl aryl chlorothiophosphates with anilines in acetonitrile at 55.0°C. The aminolyses of aryl phenyl chlorothiophosphates and aryl 4-chlorophenyl chlorothiophosphates with X-anilines in acetonitrile at 55.0°C are studied. The cross-interaction constants, ρXY, are negative for both substrates. The obtained kinetic isotope effects (kH/kD) involving deuterated aniline (XC6H4ND2) nucleophiles are greater than unity, kH/kD> 1, suggesting that the rate-determining step involves partial deprotonation of the aniline by hydrogen bonding. This is in line with a front-side attack concerted mechanism through a hydrogen-bonded four-center-type transition state. On the basis of the cross-interaction constant and primary kinetic isotope effects, authors proposed a concerted SN2 mechanism with front- and back-side nucleophilic attack on substrate. A hydrogen-bonded, four-center TS is suggested for a front-side attack, while the TBP-5C TS is suggested for a back-side attack. The MO theoretical calculations of the model reactions of substrates with ammonia nucleophile are carried out. The charge densities calculated by the NPA18 are more negative and more positive for the leaving group (Cl) and the nucleophile, respectively, in the front-side attack TS than in the back-side attack TS, indicating that the degree of stabilization by specific solvation effects could be larger in the front-side attack TS. This is supported by the results of NBO analysis at the B3LYP/ 6-311+G(d,p) level and the calculated activation energy barriersat the CPCM-MP2/6-31+G(d) level of theory. Buncel et al. [2a] reported the nucleophilic displacement at the phosphorus center of [O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothiate, by oxygen nucleophiles in aqueous solution at 25.0°C. In this paper authors concluded that the reactions between the substrate and oxygen nucleophioles proceed through concerted mechanism for nucleophilic attack at the phosphorus center. This conclusion was made based on the Brönstedplots, logkNuvs. pKa of nucleophilies shows a linear plot for the series of structurally related phenoxides in the pKa ranges 5.4-10.0, but curve obtained in the highly basic region corresponding to CF3CH2O- and HO- as nuleophiles. The βNu value was 0.49 ± 0.01 (R2 = 0.998). The linearity of the plot indicates a concerted mechanism for nucleophilic attack at the phosphorus center of the substrate. The logklgvs. pKa gave βlg = -0.39 ± 0.04 (R2 = 0.973). The combined values of βNu and βlg gave βeq= 0.88 (βNu– βlg) indicates that the TS for the symmetrical reaction has no significant phosphorylium ion character.
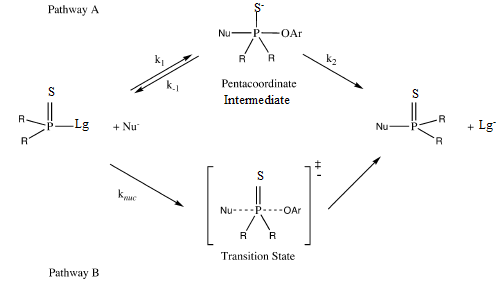 | Scheme 1. Alternative Mechanisms, an Addition-Elimination (AN + DN) Pathway with the Formation of a Pentacoordinate (Phosphorane) Intermediate (Pathway A) and a Concerted (ANDN) Reaction (Pathway B) |
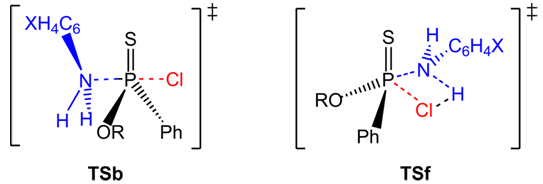 | Figure 1. Backside Attack In-Line-Type TSb and Front-Side Attack Hydrogen Bonded, Four-Center-Type TSf |
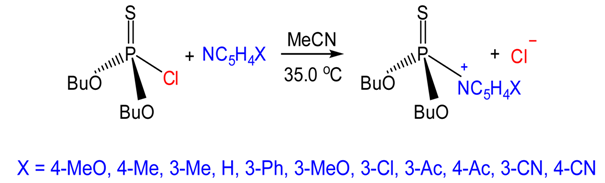 | Figure 2. The Reactions of Dibutylchlorothiophosphate with X-pyridines in MeCN at 35.0C |

2.2. Stepwise Mechanism
- Harger et al. [10] reported that the reactions between thiophosphinyl and thiophosphonyl chlorides and R2NH proceed through stepwise mechanism forming thiophosphene intermediates. Authors reported that the substitution rates increased by factors of 80 and >103 respectively when the reactions of ArCH2P(S)(Ph)Cl and ArCH2P(S)(NMe2Cl) with Et2NH, changing ArCH2 from benzyl to 4-nitrobenzyl were studied. Authors proposed the formation of intermediate by elimination-addition mechanism based on following grounds. Authors observed that in the course of reactions the ability reduced markedly to discriminate between competing Et2NH and Me2NH nucleophiles. When the substrates reacted with Et2ND, the nitrobenzyl substrates gave deuterium containing products in the benzylic methylene group.Lee et al. reported [11] the kinetic studies on the reactions of ethyl methyl and ethyl propyl chlorothiophosphates with X-pyridines have been carried out in acetonitrile at 35.0C. The substituent effects of the nucleophiles upon the pyridinolysis rates correlate with those for a typical nucleophilic substitution reaction where the stronger nucleophile leads to a faster rate with a positive charge development at the nucleophilic N atom in the transition state (TS). However, both the Hammett (log k2vs.σX) and Brönsted [log k2vs.pKa(X)] plots were biphasic concave upwards with a break point at X = H) and 3-Ph, respectively. The rate of X = H is slightly faster than that of X = 3-Ph. The magnitudes of ρX [= −7.27 and −6.96] and βX [= 1.50, 1.44] with strongly basic pyridines are 3-4 times larger than those [ρX = –2.54, –2.16; βX = 0.43, 0.36] with weakly basic pyridines.
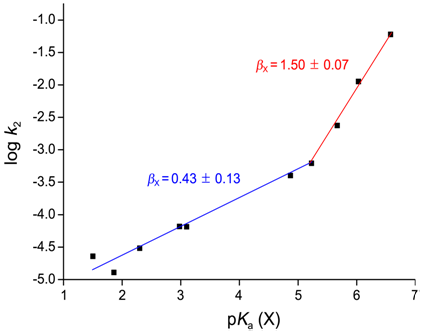 | Figure 3. Brönsted Plot of the Reactions of Ethyl Methyl Chlorothiophosphate with X-Pyridines in MeCN at 35.0C |
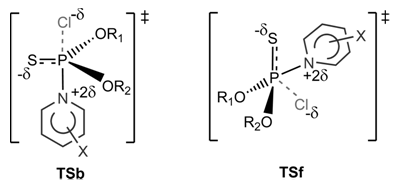 | Figure 4. Backside Attack TSb and Frontside Attack TSf |
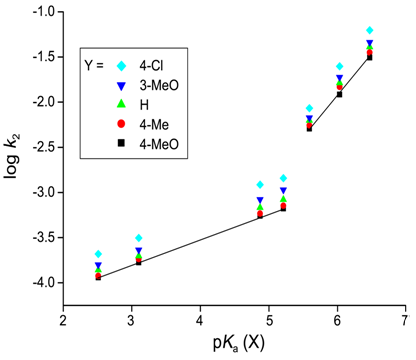 | Figure 5. Brönsted Plots with X of the Reactions of Y-Aryl Propyl Chlorothiophosphates with X-Pyridines in MeCN at 35.0C |
2.3. Mechanism Change
- Guha et al. [14] reported a mechanism change at thiophosphoryl center. Authors reported concurrent primary and secondary deuterium kinetic isotope effects in reaction mechanism change in reactions of O-aryl methyl phosphonochloridothioates with anilines. In the course of reactions the cross-interaction constants were negative (ρXY(H) = -0.95 and ρXY(D)= -1.11) for stronger nucleophiles, while positive (ρXY(H) =+0.77 and ρXY(D)= +0.21) for weaker nucleophiles. These kinetic results indicate that the mechanism changes from a concerted process involving front side nucleophilicattack for stronger nucleophiles to a stepwise process with a rate limiting leaving group expulsion from the intermediate involving backside attack for weaker nucleophiles. A hydrogen-bonding, four-center-type transition state TS was suggested for a front side attack, while a trigonal bipyramidal pentacoordinate TS was suggested for a backside attack. The unusually small deuterium kinetic isotope effects; as small or equal to 0.4, for weaker nucleophilies seem to be ascribed to serve steric congestion in the Transition State (TS).
 | Figure 6. Reaction System of Pyridinolysis of Thiophosphinic Chlorides |
3. Conclusions
- Organothiophosphorous compounds are useful in agricultural, pharmaceuticals and Textile chemicals. A significant number of works have been reported on thiophosphoryl (P=S) transfer reactions. Because predicting the effects of replacing the oxygen atom in the phosphoryl group (P=O) with sulfur is necessary. This review article includes summary of results of published papers from 1994-2014. There are two main types of mechanisms at P=S center have been reported in literature: concerted involving displacementat phosphorus through a single pentacoordinate transition state (TS) and stepwise mechanism involving a trigonal bipyramidal pentacoordinate (TBP-5C) intermediate. In some cases mechanistic change from concerted to stepwise or vice versa has been reported.
References
| [1] | [a] Quin L. D., Quin G. S. “A Guide to Organophosphorus Chemistry.” Wiley: New York (2000) Chapter 11. [b] Engel R., Ed. “Handbook of Organophosphorus Chemistry.” Marcel Dekker. New York (1992) p 465. [c] Hoque M. E. U., Dey S., Guha A. K., Kim C. K., Lee, B. S. and Lee H. W. “Kinetics and Mechanism of the Aminolysis of Aryl Phenyl Chlorothiophosphates with Anilines.”J. Org. Chem 72 (2007) 5493-5499. |
| [2] | [a] Omakor J. E., Onyido I., Vanloon G. W. and Buncel E.J. Chem. Soc.Perkin Trans. 2, (2001) 324. [b] Onyido I., Swierczek K., Purcel J. and Hengge A. C.J. Am. Chem. Soc., 127 (2005) 7703. |
| [3] | Barai H. R., Hoque M. E. U., Lee M. and g Lee H. W. “Kinetics and Mechanism of the Anilinolyses of O-Methyl, O-Propyl and O-IsopropylPhenyl Phosphonochloridothioates in Acetonitrile”. Bull. Korean Chem. Soc. Vol. 34 (2013) 1096-1099. |
| [4] | [a] Hoque, M. E. U., Lee, H. W. Bull. Korean Chem. Soc. 33(2012) 2707. [b] Adhikary, K. K., Lumbiny, B. J., Dey, S., Lee, H. W. Bull. Korean Chem. Soc.32 (2011) 2628. [c] Lumbiny, B. J., Lee, H. W. Title. Bull. Korean Chem. Soc.29 (2008) 2065. |
| [5] | Hoque M. E. U. and Lee H. W. “Pyridinolysis of Dibutyl Chlorothiophosphate in Acetonitrile”. Bull. Korean Chem. Soc. Vol. 33(2012) No. 3.1085-1088. |
| [6] | Hehre, W. J.; Random, L.; Schleyer, P. V. R.; Pople, J. A. “Ab InitioMolecular Orbital Theory”; Wiley: New York, 1986; Chapter 4. |
| [7] | (a) Taft, R. W. Steric Effect in Organic Chemistry, Newman, M.S., Ed.; Wiley: New York, 1956; Chapter 3. (b) Exner, O. Correlation Analysis in Chemistry: Recent Advances; Chapman, N. B., Shorter, J., Eds.; Plenum Press: New York, 1978; p 439. |
| [8] | (a) Lee, I. Chem. Soc. Rev. 1990, 19, 317. (b) Lee, I. Adv. Phys.Org. Chem. 1992, 27, 57. (c) Lee, I.; Lee, H. W. Collect.Czech. Chem. Commun. 1999, 64, 1529. |
| [9] | Ik-Hwan Um, Kalsoom Akhtar, Young-Hee Shin, and Jeong-Yoon Han, “Aminolyses of Aryl Diphenylphosphinates and Diphenylphosphinothioates: Effect of Modification of Electrophilic Center from P=O to P=S”, J. Org. Chem.2007,72, 3823-3829. |
| [10] | M. P. Cooggan and M. J. P. Harger, “Nucleophilic substitution in benzylicthiophosphinyl and thiophosphonyl chlorides: the contribution of elimination–addition pathways with methylenethioxophosphorane (thiophosphene) intermediates”, J. Chem. Soc. Perkin Trans. 2, 1994, 2101. |
| [11] | Hasi Rani Barai and Hai Whang Lee, “Kinetics and Mechanism of Pyridinolyses of Ethyl Methyl and Ethyl Propyl Chlorothiophosphates in Acetonitrile”, Bull. Korean Chem. Soc. 2013, Vol. 34, No. 11, 3372-3375. |
| [12] | Hasi Rani Barai and Hai Whang Lee, “Pyridinolyses of O-Propyl and O-Isopropyl Phenyl Phosphonochloridothioates in Acetonitrile”, Bull. Korean Chem. Soc. 2013, Vol. 34, No. 9 2811-2814. |
| [13] | Hasi Rani Barai and Hai Whang Lee, Kinetics and Mechanism of Pyridinolyses of Aryl Methyl and Aryl Propyl Chlorothiophosphates in Acetonitrile, Bull. Korean Chem. Soc. 2014, Vol. 35, No. 2 483-488 |
| [14] | Concurrent Primary and Secondary Deuterium Kinetic Isotope Effects in Anilinolysis of O-Aryl Methyl Phosphonochloridothioates. Md. EhteshamUlHoque, Arun Kanti Guha, Chan Kyung Kim, Bon-Su Lee and Hai Whang Lee, Org. Biomol. Chem. 2009, 7, 2919-2925. |
| [15] | Md. EhteshamUlHoque, Nilay Kumar Dey, ArunKantiGuha, Chan Kyung Kim, Bon-Su Lee, and Hai Whang Lee, “Kinetics and Mechanism of the Pyridinolysis of Diphenyl Phosphinic and Thiophosphinic Chlorides in Acetonitrile”, Bull. Korean Chem. Soc. 2007, Vol. 28, No. 10, 1797-1802. |