-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2015; 5(1): 19-22
doi:10.5923/j.chemistry.20150501.03
Electric Hindrance and Dipole Moments in 2-Aminopyridine Nitration
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFrancisco Sánchez-Viesca, Reina Gómez
Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico, México D.F., México
Correspondence to: Francisco Sánchez-Viesca, Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico, México D.F., México.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
The different isomer yields observed in many aromatic electrophilic substitution reactions can be explained by steric hindrance. However, this is not the case when there are drastic differences in the reaction yields of the isomeric products obtained. This is generally due to the presence of other factors, for instance, electric rejection between two positive charges in the reaction stage. Thus, a very important point to bear in mind is electric hindrance, a new theoretical concept. We have taken as an example 2-aminopyridine nitration. We provide an extended theory on this subject, which is in accord with the observed regiochemistry and with the reaction yields of the isomeric products obtained. Dipole moments were also taken into account. We discuss too the 2-nitraminopyridine rearrangement in acidic medium. The theoretical discussion is also in accord with reported trans-nitration experimental results. Our proposals were contrasted too with the findings from thermolysis and photolysis experiments carried out with 2-nitraminopyridine.
Keywords: Electric hindrance, Nitration, Reaction mechanisms, Reactive intermediates, Regioisomers
Cite this paper: Francisco Sánchez-Viesca, Reina Gómez, Electric Hindrance and Dipole Moments in 2-Aminopyridine Nitration, American Journal of Chemistry, Vol. 5 No. 1, 2015, pp. 19-22. doi: 10.5923/j.chemistry.20150501.03.
1. Introduction
- Electric Hindrance is a new concept in Organic Chemistry. It has been introduced by us and recognized by the American Chemical Society [1]. In this communication, this theoretical concept has been used to explain the remarkable differences shown in the regioisomers yields obtained in 2-aminopyridine nitration. Other theoretical concepts were also employed. A causal relationship was found on this subject after a careful analysis of the electronic and electric effects present both in the substrate as well on the reaction intermediates in 2-aminopyridine nitration.The rearrangement of 2-nitraminopyridine to amino-nitropyridines has also been studied. Our proposal on the rearrangement type is in accord with trans-nitration experiments carried out by other authors, as will be discussed later.
2. Theoretical Part
- There are several reviews on aminopyridines [2-6]. An interesting reaction is 2-aminopyridine nitration due to the contrasts observed in its regiochemistry.When Tchitchibabin nitrated 2-aminopyridine [7, 8], obtained as main product 2-amino-5-nitropyridine, 1, and 2-amino-3-nitropyridine, 2, as by-product (Figure 1). Since the Tchitchibabin papers are written in Russian and the published English and French abstracts are short, there are later communications, English [9] and American [10], on 2-aminopyridine nitration. These are detailed procedures based on the original experiments.
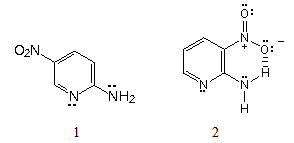 | Figure 1. Dissimilar reaction products from 2-aminopyridine nitration |
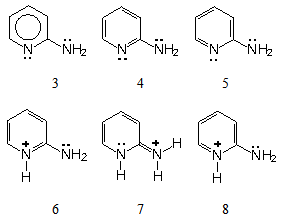 | Figure 2. Non equivalent 2-aminopyridine representations due to its reactivity, plus derived cations |
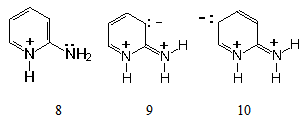 | Figure 3. High energy resonance structures of 2-aminopyridinium cation |
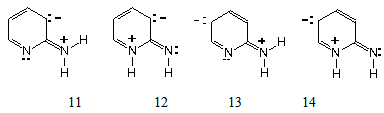 | Figure 4. Neutral dipole structures as reactive intermediates |
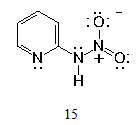 | Figure 5. 2-Nitraminopyridine, kinetic product in 2-aminopyridine nitration |
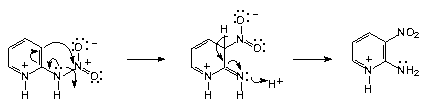 | Scheme 1. An erroneous reaction mechanism that has been proposed in order to explain the 2-nitraminopyridine rearrangement |
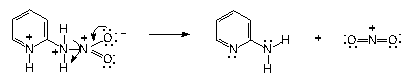 | Scheme 2. Fragmentation mechanism of 2-nitraminopyridine in acidic medium and higher temperature (40-50°C) |
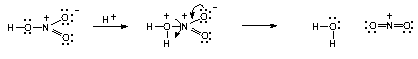 | Scheme 3. Molecular fragmentation due to contiguous positive charges, as proposed in Scheme 2 |
3. Conclusions
- In order to explain the experimental facts, i.e., the regiochemistry and the reaction yields in 2-aminopyridine nitration, a novel theory has been provided. Our study includes a discussion on the type and course of 2-nitraminopyridine rearrangement. We have deduced reasonable grounds for the studied chemical reactions. Our proposals are in complete agreement with all the available experimental results.The outstanding points are:1. Steric hindrance is not the determining factor in the regiochemistry of 2-aminopyridine nitration.2. Electric hindrance, due to repulsion between two positive charges, is the directing factor in the above reaction.3. 2-Nitraminopyridine is the kinetic product in 2-aminopyridine nitration.4. The regioisomers resulting from ring nitration are the thermodynamic reaction-products.5. 2-Nitraminopyridine rearrangement does not occur via a cyclic reaction mechanism, in a chain sequence, as has been suggested previously.6. The above mentioned rearrangement proceeds via protonation, dissociation and ring-nitration, i.e., it is an intermolecular pathway.