-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2015; 5(1): 1-9
doi:10.5923/j.chemistry.20150501.01
Effect of Decomposition Temperature on the Crystallinity of α-Fe2O3 (Hematite) Obtained from an Iron(III)-Hexamethylenetetramine Precursor
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLDivine Mbom Yufanyi1, Agwara Moise Ondoh2, Josepha Foba-Tendo3, Ketcha Joseph Mbadcam2
1Department of Chemistry, Faculty of Science, The University of Bamenda, Bambili, Bamenda, Cameroon
2Department of Inorganic Chemistry, Faculty of Science, University of Yaounde I, Yaounde, Cameroon
3Department of Chemistry, Faculty of Science, University of Buea, Buea, Cameroon
Correspondence to: Josepha Foba-Tendo, Department of Chemistry, Faculty of Science, University of Buea, Buea, Cameroon.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Iron(III) oxides has often been synthesized by techniques that require expensive equipment, extra purification steps and long reaction times. For practical applications, methods that use readily available, cost-effective and non-toxic precursors should be employed. Nanoparticles of α-Fe2O3 were synthesized by thermal decomposition of a precursor prepared from hexamethylenetetramine (HMTA) and iron(III) nitrate in an ethanol/water mixture. The precursor, [Fe(HMTA)2(H2O)3(NO3)](NO3)2·2H2O, was characterized by elemental analysis, Fourier transform infrared spectroscopy (FTIR), and thermal gravimetric analysis. It was calcined at 300, 400 and 500ºC for 2 h, and the iron oxide obtained was characterized by X-ray diffraction (XRD), FTIR, scanning electron microscopy, high resolution transmission electron microscopy and nitrogen physisorption. XRD shows that the α-Fe2O3 obtained changed from amorphous to crystalline with variation in calcination temperature. The particles tend to form a gel-like porous matrix. Our approach uses simple and cheap precursors, which should make it suitable for large-scale synthesis.
Keywords: Iron Oxide, Nanoparticles, Hematite, Thermal Decomposition, Hexamethylenetetramine
Cite this paper: Divine Mbom Yufanyi, Agwara Moise Ondoh, Josepha Foba-Tendo, Ketcha Joseph Mbadcam, Effect of Decomposition Temperature on the Crystallinity of α-Fe2O3 (Hematite) Obtained from an Iron(III)-Hexamethylenetetramine Precursor, American Journal of Chemistry, Vol. 5 No. 1, 2015, pp. 1-9. doi: 10.5923/j.chemistry.20150501.01.
Article Outline
1. Introduction
- Transition metal oxide nanoparticles are of scientific and technological importance because of their size-dependent physico-chemical properties and their potential applications in the fields of catalysis, electronics, energy storage, gas detection and magnetic resonance imaging [1-5]. Among these oxides, iron(III) oxide is of particular interest. Iron(III) oxide is polymorphic in nature with the α- and γ-polymorphs occurring in nature as the minerals hematite and maghemite, respectively [6, 4, 7-9]. The β- and ε-polymorphs, as well as the amorphous phase have been reported [6, 7, 10]. Hematite (α-Fe2O3) has a hexagonal unit cell in which two-thirds of the octahedral sites are occupied by Fe3+ ions (corundum structure) [11]. Hematite (α-Fe2O3) is an n-type semiconductor (Eg = 2.1 eV) as well as an interesting anode material which has been investigated for a wide range of applications because of it is non-toxic, readily available, and high resistance to oxidative change [3, 9, 11]. It has found applications in the manufacturing of gas sensors [12], catalysts and photocatalysts [13-15], magnetism [16, 17], lithium ion batteries [1] and electrochemical capacitor [18]. Iron(III) oxides (hematite in particular) with different particle sizes and morphologies have been obtained by a variety of physical and chemical approaches, such as chemical precipitation [18], solvothermal [19, 17], pulsed layer ablation [20], electro-spinning [1], hydrothermal [9, 11, 21], and sol-gel methods [4]. Most of these techniques require expensive equipment, extra purification steps and long reaction times. For practical applications, the synthesis should be based on readily available, non-toxic and cheap precursors, as well as simple synthetic procedures without the necessity for additional purification steps. The synthesis of α-Fe2O3 nanoparticles by thermal decomposition of organo-iron compounds or iron complexes has also been reported [22-24, 15, 25, 5, 26, 27]. By proper choice of the precursor and the calcination conditions, this could be a simple and cost-effective technique for the preparation of oxide particles with controlled morphologies. Hexamethylenetetramine (HMTA) is a cheap and readily available heterocyclic organic compound with a cage-like structure similar to adamantane. It is highly soluble in water and polar organic solvents. HMTA is a versatile ligand that can serve as a terminal monodentate or as bi-, tri- and tetradentate bridging ligand [28]. Apart from coordinate bonds, HMTA can also (depending on the conditions of synthesis and the solvent used) be involved in the formation of hydrogen bonds [29, 28]. The kinetics and thermodynamics of the thermal decomposition of such H-bonded transition metal species, leading to the formation of metal oxides (Mn, Ni, Zn, Cd), metal nitrides, or metal nanoparticles (Ni, Co; Ni-Mo and Co-Mo carbides) in a carbon matrix, have already been reported [30-34]. In this paper we report the synthesis and characterization (morphology and surface area) as well as the effect of calcination temperature on the crystallinity of α-Fe2O3 nanoparticles obtained by the thermal decomposition of an iron(III)-HMTA precursors.
2. Materials and Methods
2.1. Chemicals
- Fe(NO3)3∙9H2O, hexamethylenetetramine and ethanol were obtained from Sigma Aldrich. The chemicals were of analytical grade and were used without further purification.
2.2. Synthesis of the Fe-HMTA Precursor
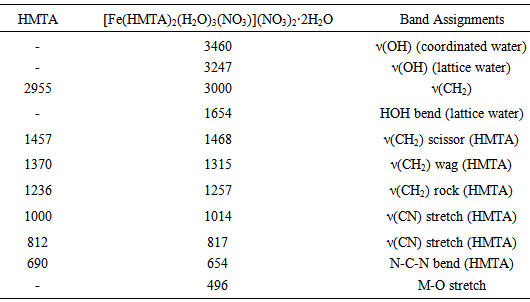
- The precursor was synthesized according to a procedure found in the literature [32]. HMTA (3 mmol, 0.421 g) was dissolved in 20 mL of ethanol/water mixture (3:1 v/v) (with sonication for 20 min at room temperature). Iron(III) nitrate (1 mmol, 0.404 g) in 10 mL of ethanol was added drop wise under magnetic stirring. The mixture was stirred for a further 2 h. The brown precipitate formed was filtered, washed several times with ethanol and dried in a desiccator over silica gel. Yield 89%; Anal. Calc. for FeC12H34N8O14: C, 23.54; H, 5.60; N, 25.16. Found; C, 23.79; H, 5.84; N, 24.82. FTIR absorption bands (cm-1): 3000-3400br, 1763w, 1654m, 1315s, 1257s, 1014s, 977m, 817m, 654m.
2.3. Synthesis of Fe2O3 Nanoparticle
- Samples of the dry precursor (0.5 g) were ground, placed in ceramic crucibles and calcined at different temperatures: 300°C (FeO-300), 400°C (FeO-400) and 500°C (FeO-500). The crucible was placed in the furnace, heated to the desired calcination temperature, and calcination in air continued for 2 h. The sample was allowed to cool down to room temperature in the furnace. The reddish powder obtained was kept in a desiccator over silica gel.
2.4. Characterization Techniques
- Elemental analysis (C, H and N) of the precursor was carried out on a Flash 2000 Thermo Scientific analyzer. FT-IR spectra were recorded from 4000 to 400 cm-1 on a PerkinElmer Spectrum Two universal attenuated total reflectance Fourier transform infrared (UATR-FT-IR) spectrometer. Thermogravimetric analysis (TGA) was obtained using a Pyris 6 PerkinElmer TGA 4000 thermal analyzer. The TGA analysis was conducted between 30 and 900°C under nitrogen atmosphere at a flow rate of 20 mL/min and a temperature ramp of 10 °C/min. The XRD diffractogram of Fe2O3 was recorded on a Bruker D8 Advance X-ray diffractometer using a Cu Kα radiation source (λ = 0.15406 nm, 40 kV and 40 mA). Scans were taken over the 2θ range from 10° to 100° in steps of 0.01° at room temperature in open quartz sample holders. The phase was identified with the help of the BrukerDIFFRACplus evaluation software in combination with the ICDD powder diffraction data base (International Centre for Diffraction Data). SEM images and EDX spectra were obtained on a JEOL JSM-7600F field-emission scanning electron microscope. Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100F microscope using a maximum acceleration voltage of 200 kV from the field emission gun. The particle size distribution was determined from the TEM image using the ImageJ software. N2-physisorption experiment for the determination of the total surface area and the average pore diameter was conducted on a Micromeritics ASAP 2020 instrument. Prior to the measurement, the sample was degassed at 200℃ for 6 h.
3. Results and Discussion
3.1. Synthesis of the Precursor
- The Fe-HMTA precursor obtained from Fe(NO3)3·9H2O and HMTA in an ethanol/water mixture (3:1 v/v) at ambient conditions in one step, is shown in scheme 1. The elemental analysis of the precursor corresponds closely to the empirical formula FeC12H34N8O14, which matches the structural formula [Fe(HMTA)2(H2O)3(NO3)](NO3)2·2H2O. Fe2O3 nanoparticles were obtained by calcination of the precursor at different temperatures. The effect of the calcination temperature on the crystallinity of the samples was investigated by several characterization techniques.
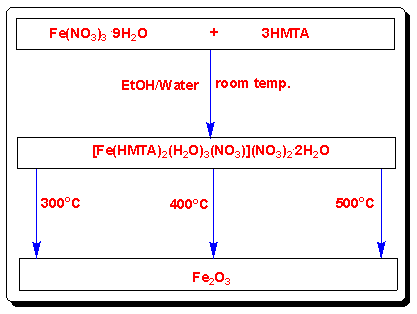 | Scheme 1. Synthesis of the Fe2O3 nanoparticles |
3.2. FTIR Analysis
- Relevant infrared bands of HMTA and the precursor complex are listed in Table 1. The FTIR spectrum of Fe-HMTA (Fig. 1) shows a broad band in the region 3247 cm-1 and a sharp peak at 1654 cm-1 which are due to ν(OH) and δ(HOH) of lattice water, while the peak at 3460 cm-1 is attributed to ν(OH) of coordinated water [35, 36, 29]. The weak and sharp band observed at 1,763 cm-1 show the coordination of a monodentate nitrate ion [29]. The band at 1238 cm-1, assigned to the C-N stretching vibration of the free HMTA, is shifted to 1257 cm-1 in the Fe-HMTA precursor, while the band at 811 cm-1, assigned to the C-N stretching vibration of HMTA is shifted to 817 cm-1 [36]. The coordination of water molecules is indicated by the IR band at 496 cm-1 [29]. The strong prominent peak at 1000 cm-1 due to the C-N stretching of HMTA is shifted to 1014 cm-1, in the Fe-HMTA precursor complex [36].In the FTIR spectrum of Fe2O3 (Fig. 1), the bands at 470 and 510 cm-1 are attributed to the Fe-O vibrations [4].
|
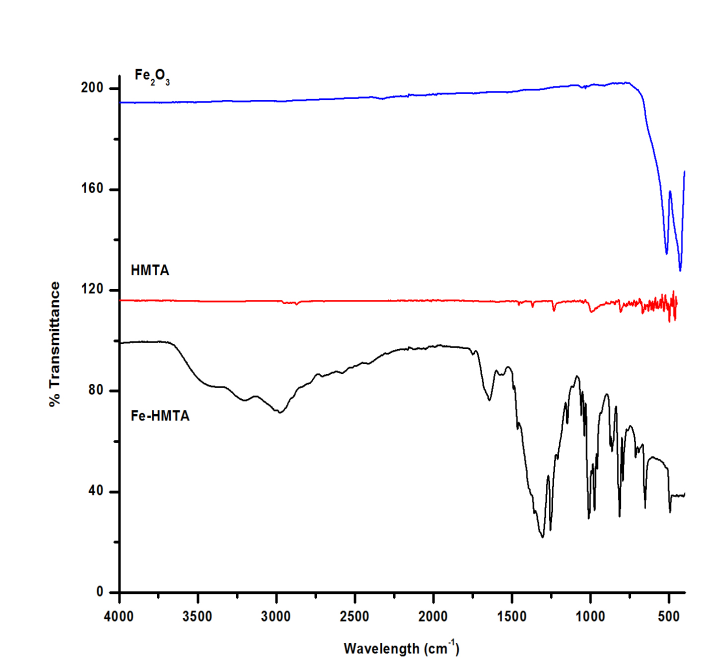 | Figure 1. FTIR spectra of Fe-HMTA precursor, HMTA and Fe2O3 |
3.3. Thermal Analysis
- The thermal behavior of the Fe-HMTA precursor is shown in Fig. 2.
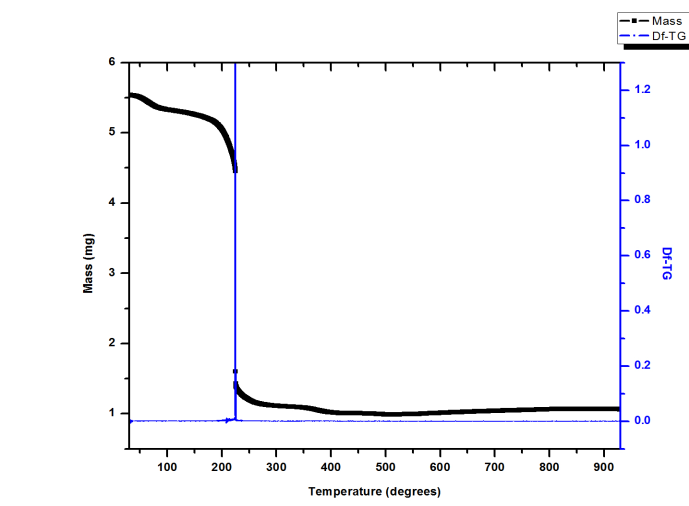 | Figure 2. TGA and Df-TG analysis of the Fe-HMTA precursor |
3.4. XRD Characterization of the Iron Oxides
- The XRD patterns of the oxides obtained are shown in Figure 3. The XRD pattern of the sample FeO-300 has low-intensity peaks indicating it is amorphous [37]. The amorphous phase crystallizes into the more stable polymorph α-Fe2O3 with increase in temperature. The samples, FeO-400 and FeO-500 have well-defined diffraction patterns with strong and sharp diffraction peaks, indicating that they are crystalline. As temperature increases, the intensity of the diffraction peaks of the samples increases and the peak width at half maximum decreases, indicating an improvement of crystallinity [15]. The peaks are indexed as the (012), (104), (110), (113), (024), (116), (122), (214) and (030) crystal planes and corresponds to the rhombohedral phase of α-Fe2O3 (JCPDS card no. 33-0664). No other impurity peaks were observed. The average crystallite sizes of the samples FeO-400 and FeO-500, calculated from the Debye-Scherrer equation [38] using the peak-width at half-height of the most intense peak (104), were found to be 45.1 nm and 71.4 nm, respectively. The increase in crystallite size with temperature is not unexpected and probably due to sintering or Ostwald ripening [39].
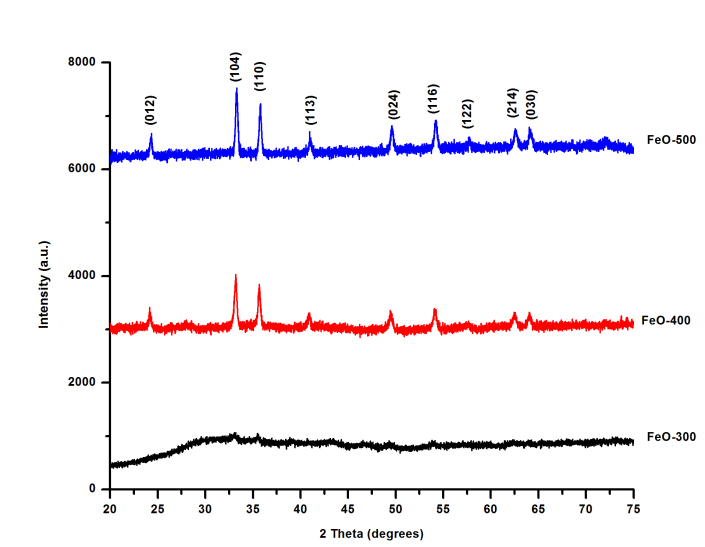 | Figure 3. XRD Pattern of α-Fe2O3 nanoparticles at different temperatures |
3.5. SEM Studies
- The SEM image (Fig. 4a) indicates that the precursor is made up of agglomerated particles having smooth surfaces, with a porous and layered morphology, while the Fe2O3 sample (FeO-500) is a highly porous solid (Fig. 4c). The surfaces of the particles are considerably coarse upon heating and an enormous number of pores develop. In FeO-500 the pores are formed by agglomerated particles that tend to form a gel-like porous matrix. This is probably caused by the rapid decomposition of the precursor at ca. 500ºC, which produces high amounts of gaseous byproducts [30]. EDX of the precursor (Fig. 4b) shows only the elements C, O, N and Fe are present. That of FeO-500 (Fig. 4d) confirms the formation of pure iron oxide as was also seen by XRD.
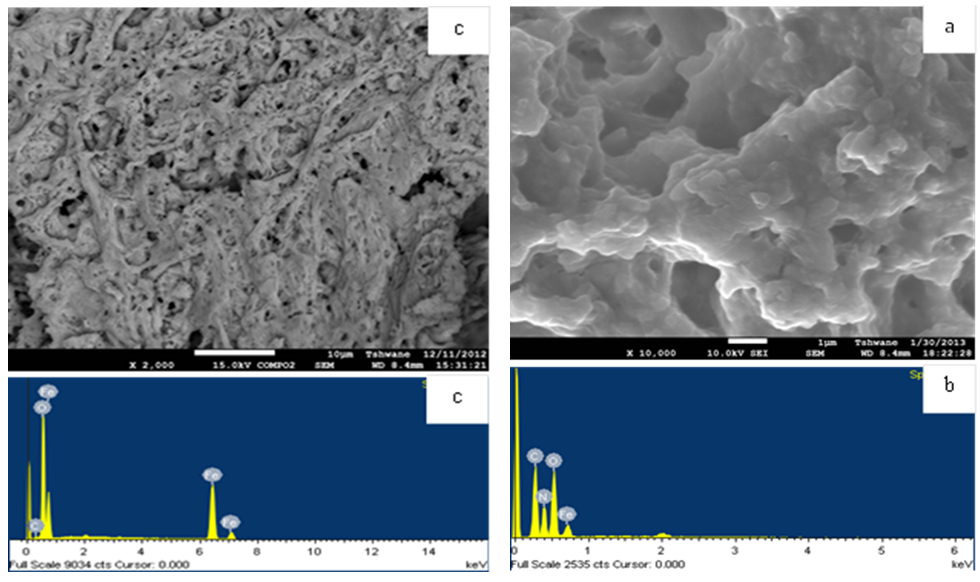 | Figure 4. (a) SEM image of Fe-HMTA precursor (b) EDX spectrum of Fe-HMTA precursor (c) SEM Image of FeO-500 (d) EDX of FeO-500 |
3.6. HRTEM Studies
- The HRTEM image (Fig. 5a) of the sample FeO-500 show particles with a hexagonal shape. The average particle diameter for FeO-500 was determined by adjusting the data obtained from the TEM images to a log normal fitting. From the resulting histogram particle sizes were in the range 40 - 81 nm, with an average particle size of 68.7 nm. This is consistent with values obtained by XRD for the crystallite sizes. The SAED image (Fig. b) shows a diffraction pattern corresponding to the polycrystalline nature of the iron oxide formed.
 | Figure 5. (a) HRTEM image of FeO-500 (b) SAED of FeO-500 |
3.7. Surface Area and Pore Size Distribution
- The surface area and pore size distribution (PSD) of FeO-500 was determined by N2 physisorption. The N2 adsorption/desorption isotherm (Fig. 6a) for FeO-500 reveals that it has a BET surface area (according to Brunauer, Emmett and Teller) 16.5 m2/g. This is probably caused by significant sintering at elevated temperature (500ºC). The Hovarth-Kawazoe (H-K) differential pore volume distribution curve (Fig. 6b) suggests that the pore diameter peaks at 1.7 nm and extends into the macroporous region.
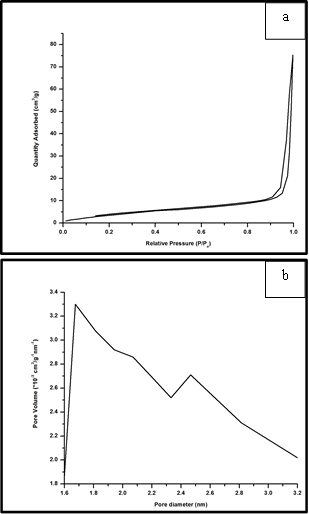 | Figure 6. (a) N2 adsorption/desorption isotherms, and (b) particle size distribution curve for the sample FeO-500 |
|
4. Conclusions
- Pure and crystalline α-Fe2O3 nanoparticles can be obtained by the thermal decomposition of a Fe-HTMA precursor at relatively low temperatures (300 – 500ºC). The sample FeO-300 is amorphous while crystallinity of the samples increases with increasing calcination temperature. The particles tend to agglomerate and form a porous gel-like matrix at high temperature. The particle size found for the sample FeO-500 (68.7 nm) compares favorably with those obtained by the decomposition of more expensive or less readily available starting materials. The sample FeO-500 has a surface area of 16.5 m2/g and an average pore diameter of 1.7 nm. This simple and cost effective low-temperature technique described is currently being extended to the synthesis of other metal oxide nanoparticles.
ACKNOWLEDGEMENTS
- The authors thank Dr. Khamlich Saleh (University of Pretoria) for assistance with the SEM and TEM images.
Conflict of Interest
- The authors declare that there is no conflict of interests regarding the publication of this paper.