-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2013; 3(5): 136-139
doi:10.5923/j.chemistry.20130305.03
Polarization by Intermolecular Induction in Pyridine N-Oxide and Its Nitration
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFrancisco Sánchez-Viesca, Reina Gómez
Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico, México, D.F., 04510, México
Correspondence to: Francisco Sánchez-Viesca, Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico, México, D.F., 04510, México.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The aim and novelty of this paper is to correct and up-date a classical theory that is two thirds wrong and besides incomplete. We mean the Linton’s theory about the pyridine N-oxide ‘excited structures’. He found that the dipole moment of pyridine N-oxide is appreciably smaller than the expected theoretical value. Thus, he postulated the contribution of three ‘excited structures’, with a negative electric charge at the 2-, 4- and 6-position. However, a typical electrophilic substitution such as nitration, afforded only the 4-nitro derivative. This discrepancy between theory and experiment prompted us to study the pyridine N-oxide physical properties, since reactivity is derived from them. Besides, these negative charged rings require an unexpected polarization and a reaction mechanism must be provided. We propose intermolecular induced polarization as a viable path, that is, a special case of electromeric effect. We uncovered that only one of the three structures before mentioned is supported by the observed reactivity as well as by 13C nuclear magnetic resonance data. Thus, we reject two of the Linton’s ‘excited structures’ and explain the regioselectivity found in pyridine N-oxide nitration.
Keywords: Autogenic electromeric effect, Induced polarization, Non-covalent interactions, Pyridine N-oxide nitration, Reactive intermediates, Resonance structures
Cite this paper: Francisco Sánchez-Viesca, Reina Gómez, Polarization by Intermolecular Induction in Pyridine N-Oxide and Its Nitration, American Journal of Chemistry, Vol. 3 No. 5, 2013, pp. 136-139. doi: 10.5923/j.chemistry.20130305.03.
Article Outline
1. Introduction
- This paper deals with the reactivity and isomeric structures of pyridine N-oxide. Since the observed reactivity in nitration experiments is not in accord with all the Linton’s ‘excited structures’ (high energy structures), Figure 1, we sought an explanation in order to obtain an agreement between theory and practice, since this is of the utmost importance.Being that the chemical properties of a compound, and therefore its reactivity, are derived from the molecular structure and the resulting physical properties (essential properties)[1], we reviewed the pyridine N-oxide references in order to deduce an experimentally based theory to explain the observed regiochemistry in the nitration reaction.
2. Experimental Facts
- Pyridine N-oxide was prepared the first time by Meissenheimer[2] from the base and perbenzoic acid (BzO2H). He described the picrate, the hydrochloride and the free N-oxide, m.p. 66-68°C, indicating it is deliquescent.Years later, Linton[3] studied the dipole moments of several amine oxides. He pointed out that pyridine N-oxide crystallizes in white needles after vacuum evaporation of a warm ether solution. The compound was kept over phosphorus pentoxide.The dipole moment was obtained determining the molar polarization of the solution measuring the dielectric constant in dilute solutions of the polar substance in a non-polar solvent (benzene). Trimethylamine oxide shows a dipole moment of 5.02 D whereas in pyridine N-oxide the dipole moment is only 4.24 D. The observed value for a semipolar bond +N ̶ O- is 4.38 D. If we add the pyridine dipole moment, pyridine N-oxide would have a 6.6 D dipole moment. In order to explain the observed small dipole, Linton proposed three ‘excited structures’, 2, 3 and 4, as contributors. Figure 1. These isomers, with the electric negative charge on the ring, change the direction of the dipole moment.
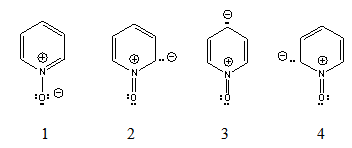 | Figure 1. Ground state and high energy or ‘excited structures’ of pyridine N-oxide |
2.1. Discussion
- As the dipole moment of pyridine N-oxide is appreciably smaller than the expected theoretical value, Linton[3] proposed the three ‘excited structures’ mentioned above (2, 3 and 4). There is no doubt about the low dipole value, since the microwave determination[10] gave a value even a little smaller. However, only the ‘excited structure’ 3 have been confirmed by 13C nuclear magnetic resonance since only the γ-carbon resonance signal appears up-field (at lower frequency). This result is in agreement with the regioselectivity observed in the pyridine N-oxide nitration experiments, as it will be discussed in the next section. Due to this concordance, we rejected the ‘excited structures’ 2 and 4 as a result of the experimental data, in accord to empiricism.Moreover, Linton did not gave an explanation as how the high-energy ‘excited structures’ can be formed. The formation of dipole-structure 3, the only acceptable one, presents theoretical problems. It is well known that the polarization of an imino group occurs towards the nitrogen atom. In the pyridine molecule, this electronic shift is the basis of Tchitchibabin’s reaction[14],[15] to obtain2-aminopyridine. This shift to the nitrogen atom is enhanced in pyridine N-oxide ground state, 1, since now there is an iminium ion, with a positive charged nitrogen atom. However, this polarization tendency must be inverted in order to form structure 2 and then 3. We can invoke the electron donor property of the negative charged oxygen towards the positive nitrogen atom to form structure 2, Figure 1. But look at the obtained adverse results: a) the nitrogen atom remains positive, since it has not been neutralized; b) a negative charge is more stable in an oxygen atom than in a carbon atom, as is now in 2; and c) the ring aromaticity has been lost.Our proposal to overcome these problems is to take into account the set of the expected resonance structures (5, 6 and 7), with normal electronic shifts. Figure 2.
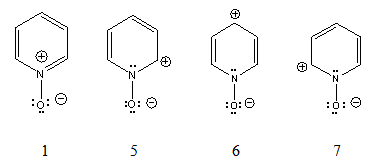 | Figure 2. Ground state and expected resonance structures of pyridine N-oxide |
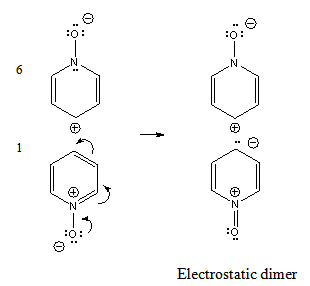 | Figure 3. Electronic shift by intermolecular induction (autogenic electromeric effect) in pyridine N-oxide |
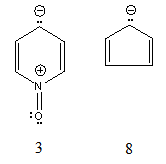 | Figure 4. Structure 3 and cyclopentadiene anion |
3. Pyridine N-oxide Nitration
- It was known that in benzene nitration the reactive species is the nitronium ion[20]. Thus, Ochiai in Japan, based on Linton’s ‘excited structures’, nitrated pyridine N-oxide in different experimental conditions[21-23]. Some years later, before the abstracts of the last two papers appeared in English, den Hertog and Overhoff in Holland[6] nitrated the same compound.Both research groups found that the reaction product is 4-nitropyridine N-oxide[24], i.e., there is regioselectivity. This result supports only one of the three structures proposed by Linton (Figure 1). It is important to note that reaction at C-2 and at C-6 would yield 2-nitropyridine N-oxide, and in a double quantity than that obtained of 4-nitropyridine N-oxide, which is not the case at all.The preparation of 4-nitropyridine N-oxide has been detailed in a Bremen University communication[25], including the IR, 1H NMR and 13C NMR spectra.
3.1. Discussion
- A comparison of the reactivity of pyridine and pyridine N-oxide has been made[26]. The N-oxide nitration (at C-4) can be effected much more readily that with pyridine, although the reaction is still more difficult than the nitration of benzene. Pyridine N-oxide is nitrated by concentrated nitric and sulphuric acids at 130°C during 3.5 hours[4] while benzene reacts with the same reagents at 50°C. Cf.[27-29].The comparison is interesting because we deduce that even structure 3, the only acceptable of Linton’s structures, is not very important. Notwithstanding the negative charge at C-4, pyridine N-oxide presents a lower reactivity than benzene, a neutral molecule, and we must take into account that the reactive species is a positive ion (nitronium ion), which would react very fast with any carbanion of Linton’s structures. So, there must be a low population of molecules having structure 3.The kinetics of nitration of pyridine N-oxide shows that the reaction occurs through the free base[30-32]. This experimental result is in accordance with theory since structure 3 derives obviously from the free base.The novel electrostatic dimer proposed in the previous discussion, yields the reactive ylide 3, and nitration occurs. The other component, structure 6, can return to the ground state, 1, then the nitronium ion originates the electromeric effect and nitration takes place.Other possibility is that remaining structures 6 can form more structures 3 from molecules in the ground state, 1, giving more electrostatic dimer.
4. Conclusions
- The formation of the ‘excited structures’ proposed by Linton for pyridine N-oxide requires a theoretical explanation since it implies an electronic shift contrary to the mesomeric effect of the iminium ion present in the ground state. In order to form the proposed ylides, the electron-donor effect of the oxygen atom must surpass the opposite mesomeric effect. Thus, a promoter is required, such as an electromeric effect. In the absence of other reactant, we explain this singularity by the participation of the resonance structure 6, with positive charge at C-4, that is, the existence of intermolecular induction.Linton’s ‘excited structures’ for pyridine N-oxide predict reaction at the 2-, 4- and 6-position. However, in an electrophilic substitution reaction such as nitration, there is reaction only at C-4 (regioselectivity), and the 13C NMR spectroscopy only supports higher electronic density at C-4. From these experimental results, we discard the ‘excited structures’ with a negative charge at C-2 and C-6, since a theory must be in assent with empirical results.We have proposed a novel push-pull mechanism via intermolecular induction, which can be named ‘autogenic electromeric effect’. We have also explained the regioselectivity of pyridine N-oxide nitration.