-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2013; 3(4): 77-95
doi:10.5923/j.chemistry.20130304.01
A Global Perspective on a Review of a Three-Year C-Nucleosides Development: 2009-2011
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLMohamed A. El-Atawy 1, 2, Mohamed Nabil Abd Al-Moaty 1, Adel Amer 1, 3
1Department of Chemistry, Faculty of Science, Alexandria University, Egypt
2Dipartimento Di Chimica, Universita Degli Studi Di Milano, Via C. Golgi 19, 20133 Milano, Italy
3Department of Applied Chemistry, Faculty of Applied Science, Taibah University, Madinah Munawwarah, Saudi Arabi
Correspondence to: Adel Amer , Department of Chemistry, Faculty of Science, Alexandria University, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
C-Nucleosides remain among the most challenging modified nucleosides to build for evaluation of their biological activities. This review includes a brief introduction of the C-Nucleosides classification and focuses on what has been done during a three year period (2009-2011) for their synthetic approaches and structural modifications. It spots locations of work and synthetic tactics, then correlate that to international vs. national transformation of methodologies. For sustainable developments science diplomacy may provide a forum for international collaborative research, which leads to intriguing topics of potential applications.
Keywords: C-nucleosides, Synthetic Strategies, Global Perspective, Science Diplomacy
Cite this paper: Mohamed A. El-Atawy , Mohamed Nabil Abd Al-Moaty , Adel Amer , A Global Perspective on a Review of a Three-Year C-Nucleosides Development: 2009-2011, American Journal of Chemistry, Vol. 3 No. 4, 2013, pp. 77-95. doi: 10.5923/j.chemistry.20130304.01.
Article Outline
1. Introduction
- Nucleosides are fundamental building blocks of biological systems. The natural nucleosides and their analogs find numerous applications as antibiotic, fungicidal, anti-tumor and anti-viral agents. Therefore, the chemistry of these compounds has been studied extensively and today nucleoside chemistry represents an important area of research for modern drug discovery. Vital elements of the structure of nucleoside are: 1) hydroxymethyl functionality; 2) spacer [e.g., sugar component] on which the hydroxymethyl group is a part; 3) heteroaryl base or aryl group attached to the sugar. C-Nucleosides are a subtype of these compounds in which the spacer and heteroaryl(aryl) groups are linked via C-C bond (Figure 1). The attractive feature of C-Nucleosides arises from the presence of this C-C glycosidic bond, which gives a greater resistance towards chemical and enzymatic hydrolysis than N-Nucleosides.
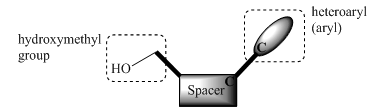 | Figure 1. Generalized structure of a C-nucleoside analogue |
2. Building up the Heterocyclic Unit on a Suitably Functionalized Carbohydrate Moiety
2.1. From Nitrile Terminal on C1 Atom
2.1.1. Cyclic C-Nucleosides
- Functionalization of the carbohydrate moiety with nitrile group at the anomeric carbon gave an important precursor for synthesis of C-Nucleoside. Protected ribofuranosyl nitrile 5a-c was converted to the corresponding ribofuranosyl thioamide (6a-c), whose reaction with ethyl bromopyruvate gave the protected tiazofurin derivative 7a-c. Treatment of the latter with methanolic ammonia afforded The thiazole C-Nucleoside 8a-c (Scheme 1). [1,
 ].
]. 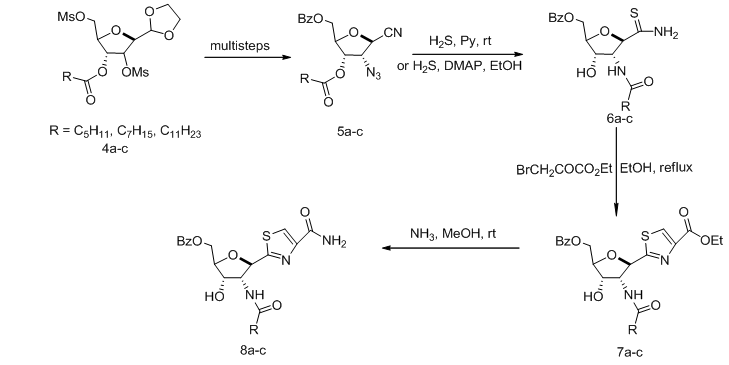 | Scheme 1. Construction of C-nucleosides 7 from nitrile precursors |
2.1.2. Homocyclic C-Nucleosides
- The homocyclic C-nucleoside analogue was also reported from the nitrile 9 using the same procedure already used for preparation of 8a-c to give the corresponding homo-C-tiazofurin analogue bearing 2,3-anhydro ribofuranosyl moiety 13 and 14, which represent the first biologically active tiazofurin analogue that demonstrate antiproliferative activity (Scheme 2). [2,
 ].
].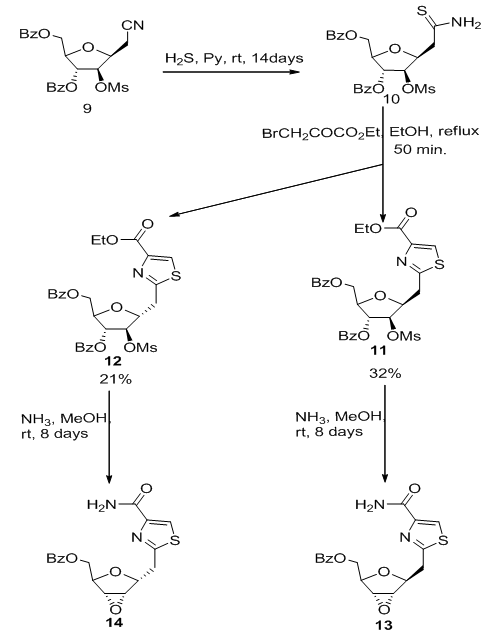 | Scheme 2. Construction of homocyclic C-nucleoside 13 from nitrile precursor |
2.2. From Alkynyl Terminal on C1 Atom
2.2.1. Cyclic C-Nucleosides
- Another essential functional group for synthesis of hetrocyclic moiety upon carbohydrate is the ethynyl group at the anomeric carbon. Sonogashira reaction of 1-ethynyldeoxyriboside 16α or 16β with trifluoroacetanilide derivatives 15a-d furnished the corresponding alkynyldeoxyribose derivatives 17α or 17β, whose cyclization led to indolyl C-Nucleoside 18α or 18β (Scheme 3). [3,
 ].
].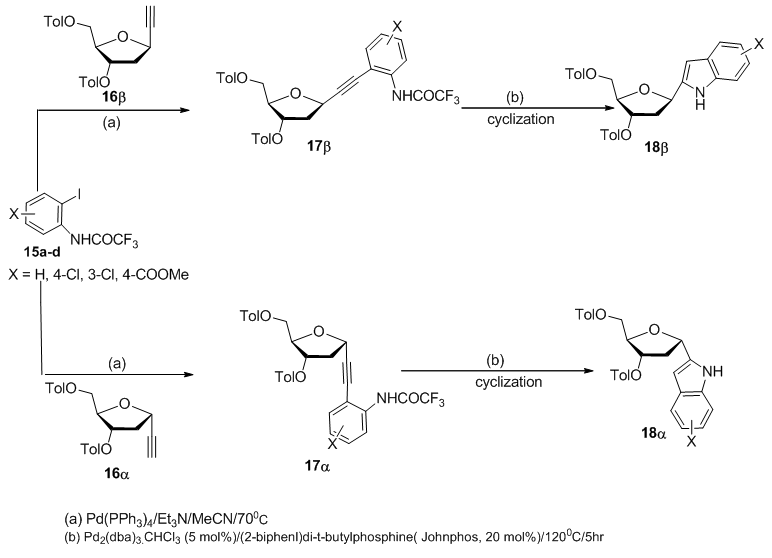 | Scheme 3. Synthetic steps to prepare C-nucleoside 18 from alkynyl precursor |
 ].
].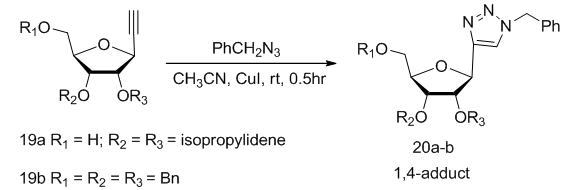 | Scheme 4. Synthesis of 1,2,3-triazole C-nucleosides 20 from alkynyl precursor |
 ] The reaction was conducted under micellar catalysis to improve the regioselctivity.
] The reaction was conducted under micellar catalysis to improve the regioselctivity. 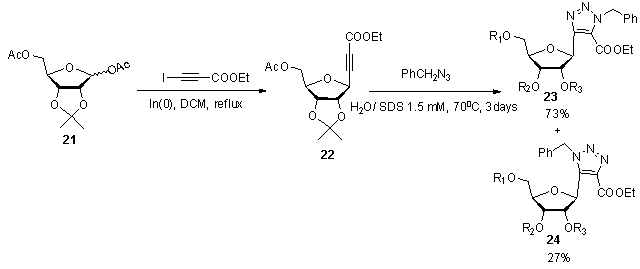 | Scheme 5. Synthetic approach to C-nucleosides 23 and 24 via 1,3-dipolar cyclization |
2.2.2. Heterocyclic C-Nucleosides
- The same methodolgy of heterocyclization of ethynyl group into 1,2,3-triazole was used to prepare the aza-C-Nucleoside analogue 28 (Scheme 6). [5,
 ,
,  ].
].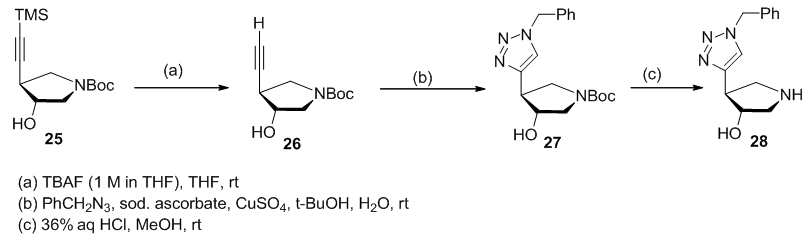 | Scheme 6. Synthetic approach to aza-C-nucleosides 28 via 1,3-dipolar cyclization |
2.3. From Carbonyl Functionality on C1 Atom
2.3.1. Cyclic and Homocyclic C-Nucleosides
- Hantzsch reaction on the aldehydo of 29a with β-ketoester 30 and enamine ester 31, under L-proline catalysis gave dihydropyridine C-Nucleoside 32a in stereoselective manner with (de 95%). The use of C-glycosyl aldehyde with elongated chain 29b-f extends the synthesis to the homo-C-Nucleoside analogue 32b-f (Scheme 7). [6,
 ,
,  ].Wittig type reaction of sugar functionalized with keto group at the 4- position 33 and stabilized phosphrane leads to 34 whose mild oxidation with pyridinium chlorochromate (PCC) afforded β-sugar- β-formy-α-β-unsaturated ester 35. Utilization of 35 in cyclocondensation reaction with hydrazine hydrate or its derivatives gave pyrazole, pyrazoline, pyridazionone pseudo C-Nucleoside (Scheme 8). [7,
].Wittig type reaction of sugar functionalized with keto group at the 4- position 33 and stabilized phosphrane leads to 34 whose mild oxidation with pyridinium chlorochromate (PCC) afforded β-sugar- β-formy-α-β-unsaturated ester 35. Utilization of 35 in cyclocondensation reaction with hydrazine hydrate or its derivatives gave pyrazole, pyrazoline, pyridazionone pseudo C-Nucleoside (Scheme 8). [7,  ].
]. 2.3.2. Acyclic C-Nucleosides
- Construction of the heterocyclic moiety on sugar can also be used for synthesis of acyclo C-Nucleoside analogs. Thus, the condensation of heterocyclic hydrazine derivatives with some monosaccharide gave the corresponding aldehydo sugar hydrazones, which on acetylation gave the o-acetylated sugar derivatives subsequent oxidative cyclization followed by de-o-acetylation afford the 1,2,4-triazole C-nucleoside. Scheme 9 [8-14,
 ,
,  ].In most cases acetylation of the sugar hydrazones (40 a, b, c, f, g) gave directly the O- acetylated cyclic C-nucleoside, on the other hand all attempts dehydrogeative cyclization of sugar hydrazone (40d) or its acetylated derivative (41d) failed. Competition between two ortho ring nitrogen (NH, =N) in the oxidative cyclization process such as in case of the O-acetylated hydrazone (41c, g, h) gave product corresponded to cyclization between the azomethine carbon and the hydrogenated nitrogen of the ring. Reaction of acid hydrazides with some aldehydo sugars gave the corresponding sugar aroylhydrazones. When those hydrazones were heated in acetic anhydride at 100 oC their corresponding 1,3,4-oxadiazoline acyclic C-nucleosides 46a-b were isolated (Scheme 10). [15-16,
].In most cases acetylation of the sugar hydrazones (40 a, b, c, f, g) gave directly the O- acetylated cyclic C-nucleoside, on the other hand all attempts dehydrogeative cyclization of sugar hydrazone (40d) or its acetylated derivative (41d) failed. Competition between two ortho ring nitrogen (NH, =N) in the oxidative cyclization process such as in case of the O-acetylated hydrazone (41c, g, h) gave product corresponded to cyclization between the azomethine carbon and the hydrogenated nitrogen of the ring. Reaction of acid hydrazides with some aldehydo sugars gave the corresponding sugar aroylhydrazones. When those hydrazones were heated in acetic anhydride at 100 oC their corresponding 1,3,4-oxadiazoline acyclic C-nucleosides 46a-b were isolated (Scheme 10). [15-16,  ,
,  ].Cyclocondensation of aldehydo sugar Schiff base 48 with thioglycolic acid in dry dioxane afforded the corresponding thiazolidinone a cyclic C-nucleoside 49 (Scheme 11). [17,
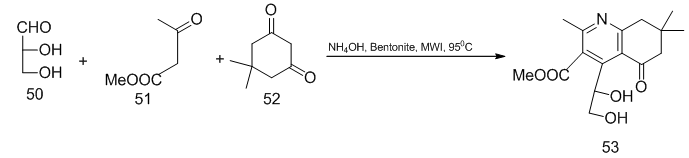
].Cyclocondensation of aldehydo sugar Schiff base 48 with thioglycolic acid in dry dioxane afforded the corresponding thiazolidinone a cyclic C-nucleoside 49 (Scheme 11). [17,  ].Following the Hantzch approach for synthesis of pyridine, treatment of D-glyceraldehyde 50 with methyl acetoacetate and dimedone 52 in presence of bentonite clay as a support , ammonium nitrate as source of ammonia, and HNO3 as oxidant was furnished, after chromatgraphic purification, C-acyclic pyridine nucleoside 53 (Scheme 12).. [18,
].Following the Hantzch approach for synthesis of pyridine, treatment of D-glyceraldehyde 50 with methyl acetoacetate and dimedone 52 in presence of bentonite clay as a support , ammonium nitrate as source of ammonia, and HNO3 as oxidant was furnished, after chromatgraphic purification, C-acyclic pyridine nucleoside 53 (Scheme 12).. [18,  ,
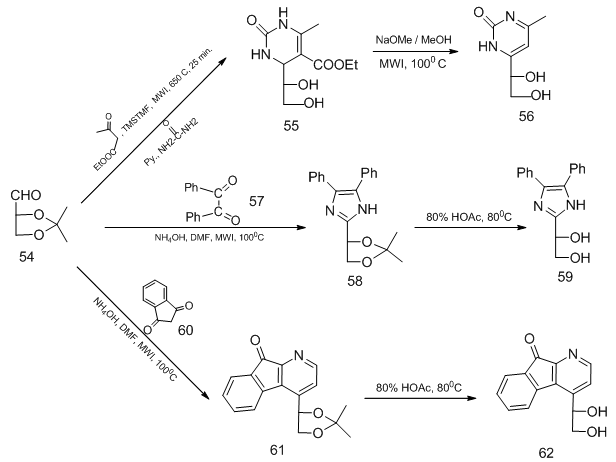
,  ]The Kidawai's method for synthesis of imidazole derivative via condensation of 1,2- dicarbonyl compound such as benzil with aldehyde in excess of ammonium hydroxide under microwave irradiation (MWI) was used for the synthesis of imidazole C-acyclic nucleoside 59 by utilizing the aldehydo functional group of protected D-glyceraldehyde 54. While condensation of 54 with ethyl acetoacetate and urea using natural phosphate doped with ZnCl2 under MWI afforded acyclic C-nucleoside having dihydropyrimidinone 55. An efficient synthesis of the C-acyclic nucleoside of indeno[1,2-b]pyridine derivative 62 was accomplished using MWI treatment of 54 with 1,3-indeno-1,3-dione in presence of ammonium acetate (Scheme 13). [18,
]The Kidawai's method for synthesis of imidazole derivative via condensation of 1,2- dicarbonyl compound such as benzil with aldehyde in excess of ammonium hydroxide under microwave irradiation (MWI) was used for the synthesis of imidazole C-acyclic nucleoside 59 by utilizing the aldehydo functional group of protected D-glyceraldehyde 54. While condensation of 54 with ethyl acetoacetate and urea using natural phosphate doped with ZnCl2 under MWI afforded acyclic C-nucleoside having dihydropyrimidinone 55. An efficient synthesis of the C-acyclic nucleoside of indeno[1,2-b]pyridine derivative 62 was accomplished using MWI treatment of 54 with 1,3-indeno-1,3-dione in presence of ammonium acetate (Scheme 13). [18,  ,
,  ]
]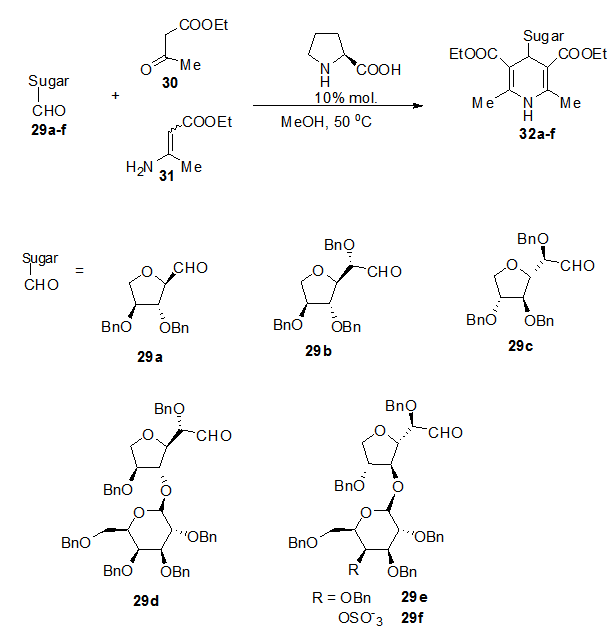 | Scheme 7. Construction of cyclic and homocyclic C-nucleosides 32 from carbonyl functionality on C1 of precursors |
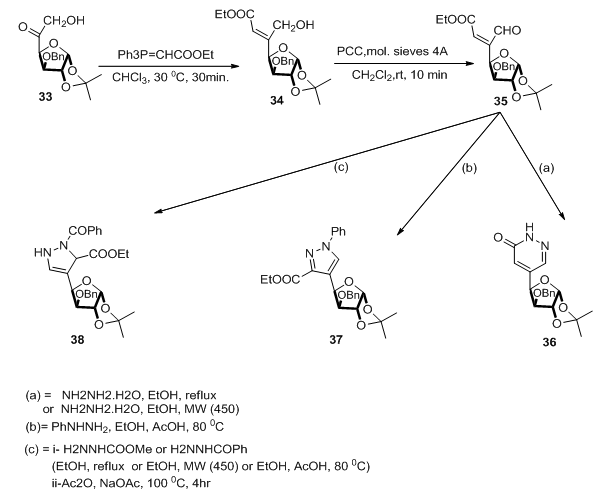 | Scheme 8. Syntheses of C-nucleosides 36-38 |
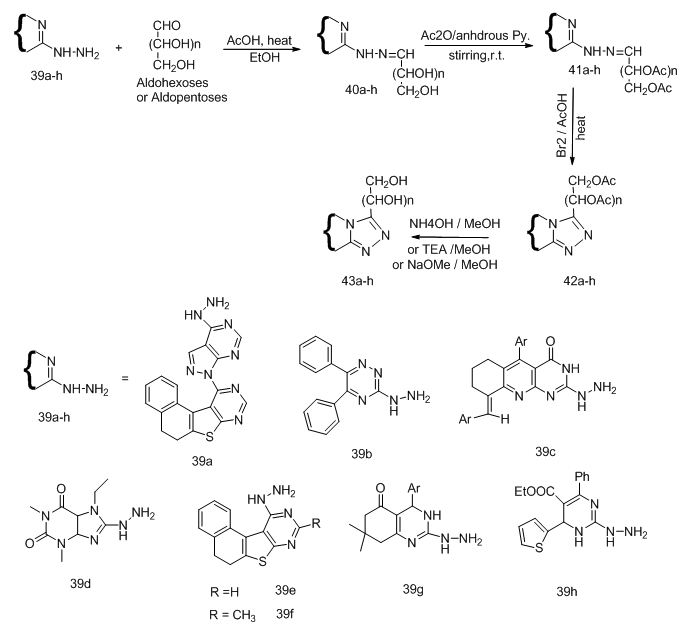 | Scheme 9. Synthetic steps for the preparations of acyclic C-nucleosides 43 from the reactions of heterocyclic hydrazines with aldoses |
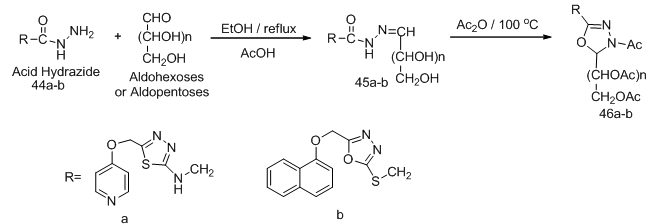 | Scheme 10. Synthetic approach to acyclic C-nucleosides 46 from hydrazides |
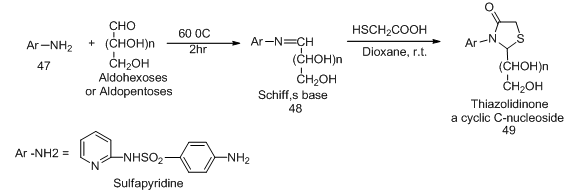 | Scheme 11. Synthetic approach to acyclic C-nucleosides 49 from aniline derivatives and aldoses |
 | Scheme 12. Synthesis of acyclic C-nucleoside 53 from aldoses via multicomponent reaction (MCR) |
 | Scheme 13. Synthetic approachs to acyclic C-nucleosides 56, 59 and 62 |
3. Direct Coupling between a Suitably Protected Carbohydrate Moiety with a Preformed Heterocyclic Compound
- The direct attachment of a preformed glycon unit to an appropriate carbohydrate is one of the most important method for synthesis of C-nucleoside. This strategy is based on ionic, free radical or metal mediated carbon-carbon bond formation. Synthesis of nucleoside derivatives of quinuclidin-3-one 63 was achieved by the reaction of 63 with D-glucose in presence of catalytic amount of zinc chloride to give the cyclic C-nucleoside analogue 64a-d. This reaction involves nucleophilic dislpacemnt of the anomeric hydroxyl group which activated by Lewis acid catalysis. On the other hand condensation of 63 with D-glucose in sodium carbonate as basic catalyst instead of Lewis acid afforded the bis- quinclidine sugar 65. In addition the Mannich base analogue 66 was synthesized by treatment of 63 with glucose and morpholine under Mannich reaction condition( Scheme 14). [19,
 ].
].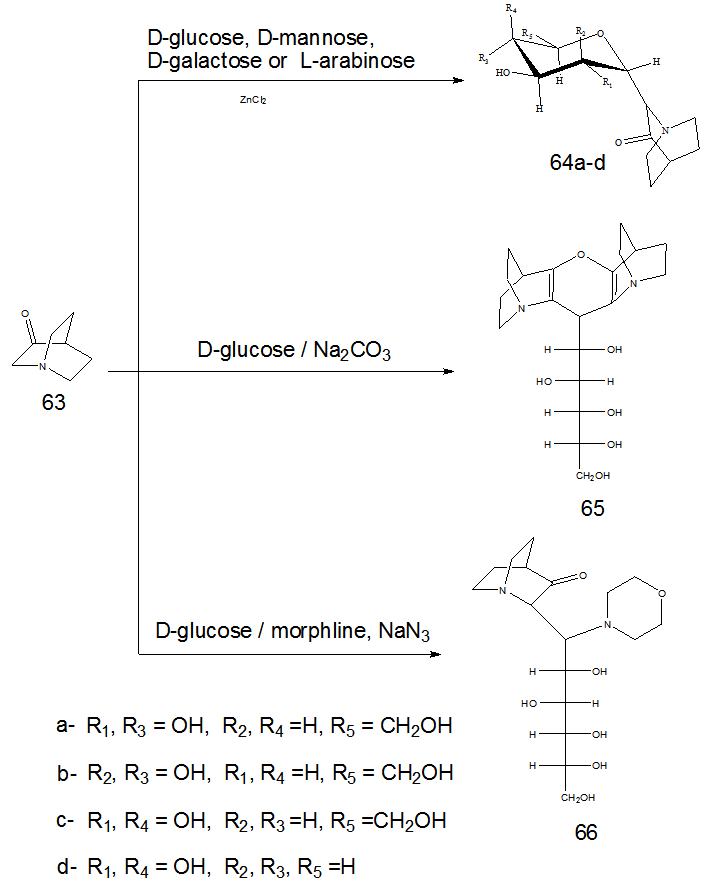 | Scheme 14. Construction of acyclic C-nucleosides 64-66 |
 ].
].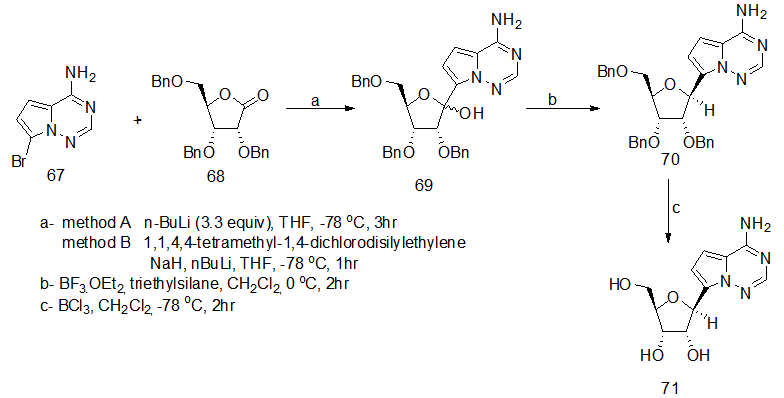 | Scheme 15. Reaction of heteroaryl lithium with sugar lactone to prepare 71 |
 ].
]. 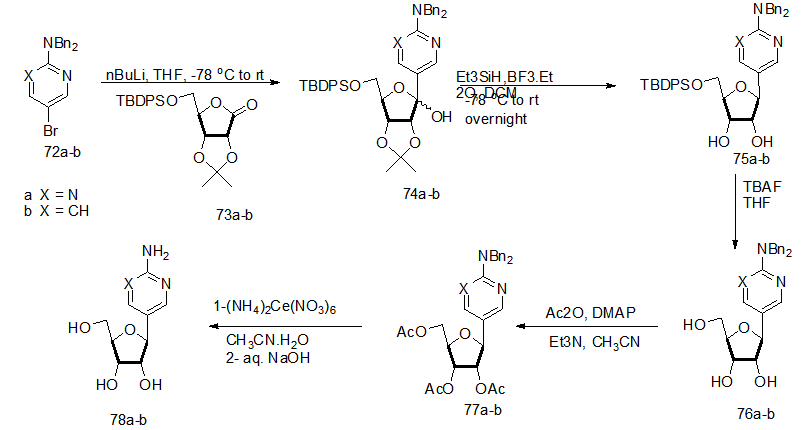 | Scheme 16. Reaction of heteroaryl lithium with sugar lactone to prepare C-nucleosides 76 |
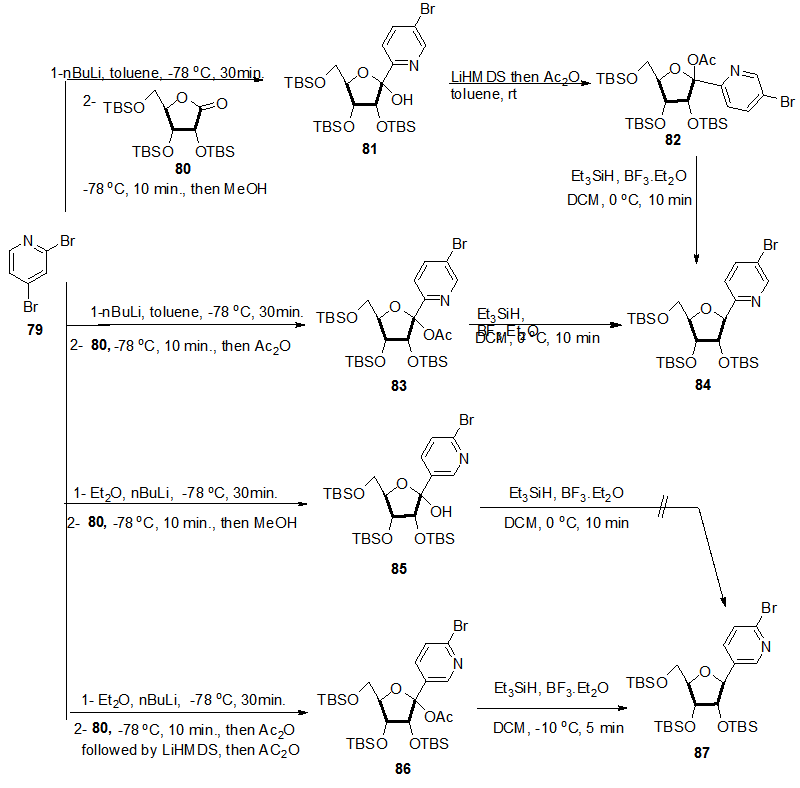 | Scheme 17. Synthetic approachs to C-nucleosides 84 and 87 from 2,4-dibromopyridine |
4. Construction of the Spacer on the Base
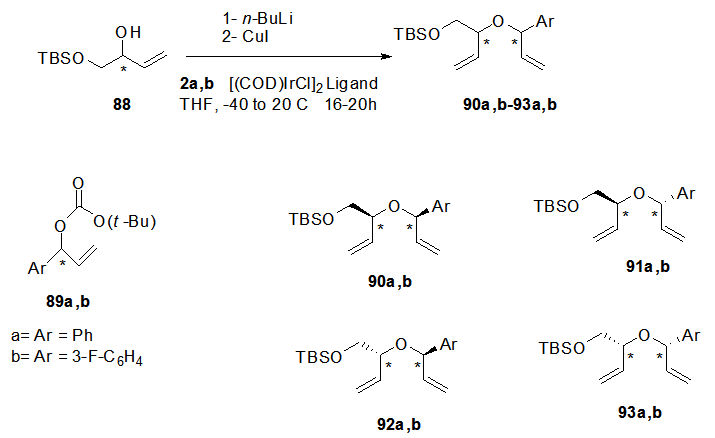
- A stereo-controlled synthesis via allylic substitution and ring closing metathesis sequence was reported to construct the sugar moiety of some aryl C-Nucleosde 108a-c. Where as the reaction of the enantiopure alcohol 88 with the enantiopure branched carbonated compounds 89a,b in presence of Ir(I) catalyst [23,
 ,
,  ] afforded the respective enantiopure products 90-93 in good yields (Scheme 18). Ring closing metathesis of the bisallyl ethers 90-93 in presence of a catalyst led to the formation of the desired 1,5-dihydrofuran derivatives 94a,b-97a,b which were deprotected by treatment with Et3N.HF to afford the corresponding alcohols 98a,b-101a,b (Scheme 19). Also vicinal dihydroxylation of the isolated cis- dihydrofuran derivatives 94a,b and 97a,b using either osmium tetroxide or ruthenium tetroxide as catalysts were performed to obtain compounds 102a,b and 103a,b in excellent yield.
] afforded the respective enantiopure products 90-93 in good yields (Scheme 18). Ring closing metathesis of the bisallyl ethers 90-93 in presence of a catalyst led to the formation of the desired 1,5-dihydrofuran derivatives 94a,b-97a,b which were deprotected by treatment with Et3N.HF to afford the corresponding alcohols 98a,b-101a,b (Scheme 19). Also vicinal dihydroxylation of the isolated cis- dihydrofuran derivatives 94a,b and 97a,b using either osmium tetroxide or ruthenium tetroxide as catalysts were performed to obtain compounds 102a,b and 103a,b in excellent yield.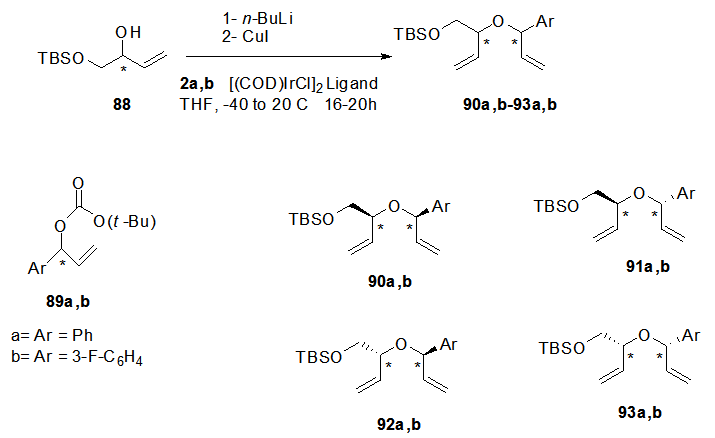 | Scheme 18. Preparation of bisallyl ethers 90-93 |
 | Scheme 19. Synthetic approaches to C-nucleosides 102 and 103 via ring closing metathesis sequence of bisallyl ethers |
 ] Thus, esterification of 104 using either acyl chlorides in CH2Cl2 in presence of Et3N and 4-dimethylaminopyridine (DMAP), or carboxylic acids, which was performed in CH2Cl2 in presence of DCC and DMAP gave the desired esters 105a-c in good yields. The obtained esters are then cyclised using MeLi in THF to obtain the cyclised lactols 106a-c which underwent reductive dehydroxylation to give the protected derivatives 107a-c followed by debenzylation using either NaI in Me3SiCl and MeCN or BBr3 in CH2Cl2 to obtain aryl C-nucleoside derivatives 108a-c.
] Thus, esterification of 104 using either acyl chlorides in CH2Cl2 in presence of Et3N and 4-dimethylaminopyridine (DMAP), or carboxylic acids, which was performed in CH2Cl2 in presence of DCC and DMAP gave the desired esters 105a-c in good yields. The obtained esters are then cyclised using MeLi in THF to obtain the cyclised lactols 106a-c which underwent reductive dehydroxylation to give the protected derivatives 107a-c followed by debenzylation using either NaI in Me3SiCl and MeCN or BBr3 in CH2Cl2 to obtain aryl C-nucleoside derivatives 108a-c.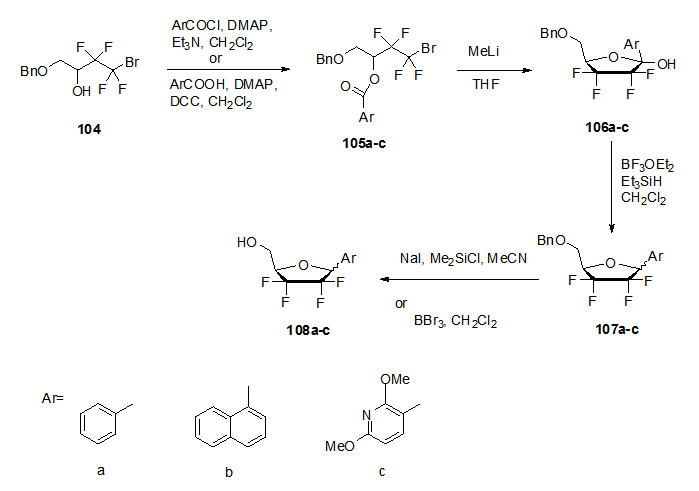 | Scheme 20. Synthetic approach to fluorinated C-nucleosides 108 |
5. Transformation of the Existing C-nucleosides to Another Ones
- Commonly transformation of the C-nucleosides to other ones is carried out to improve the biological activity of the existing C-nucleosides without breaking the C-C bond between both the sugar and the base moieties.Modification of the existing C-nucleosides occurred either in the sugar or base parts of the existing natural or synthetic C-nucleosides.
5.1. Modifications on the Base Moiety
- Modifications on the heteroaryl (aryl)-C-nucleosides using organometallic catalysis were reported. In 2009 Hocek et. al. described the aminocarbolylation of bromophenyl- C-ribonucleoside 109 with CO (1 atm) with different amines in presence of lead acetate, xantphos and potassium phosphate in toluene to give the corresponding amides 110a-c (Scheme 21, Table 1). [25,
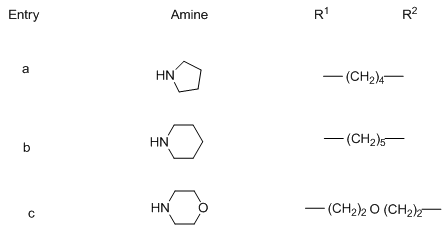
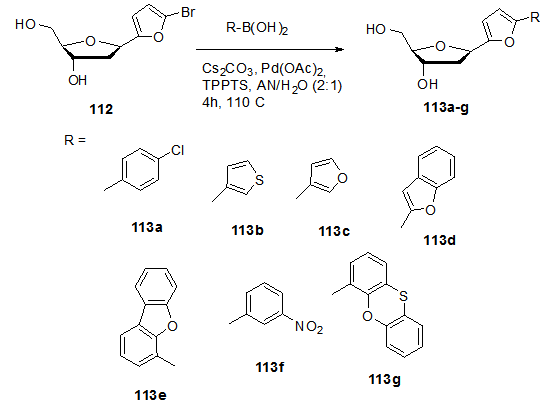
 ] Deprotection of the silylated nucleosides 110a-c using Et3N.3HF at 40°C for two days followed by treatment with K2CO3 led to the formation of the desired free C-ribonucleosides 111a-c. Benzamide-C-ribonucleosides were found to have a biological character as to be a strong cytostatic agent including apoptosis in cancer cells.The unprotected 5-bromofuran-C-nucleoside 112 was used as a potential intermediate for aqueous phase cross coupling reactions. The Suzuki-Miyaura cross coupling reaction of this compound with different aryl boronic acids carried out in presence of lead acetate,tris(3-sulfophenyl)phosphine trisodium salt (TPPTS) ligand and Cs2CO3 as a base for 4h at 120°C yielded the biaryl β-C-nucleosides 113a-g (Scheme 22). [26,
] Deprotection of the silylated nucleosides 110a-c using Et3N.3HF at 40°C for two days followed by treatment with K2CO3 led to the formation of the desired free C-ribonucleosides 111a-c. Benzamide-C-ribonucleosides were found to have a biological character as to be a strong cytostatic agent including apoptosis in cancer cells.The unprotected 5-bromofuran-C-nucleoside 112 was used as a potential intermediate for aqueous phase cross coupling reactions. The Suzuki-Miyaura cross coupling reaction of this compound with different aryl boronic acids carried out in presence of lead acetate,tris(3-sulfophenyl)phosphine trisodium salt (TPPTS) ligand and Cs2CO3 as a base for 4h at 120°C yielded the biaryl β-C-nucleosides 113a-g (Scheme 22). [26,  ] These nucleosides have high fluorescence properties that can be used as fluorescent labeling in biomolecules.
] These nucleosides have high fluorescence properties that can be used as fluorescent labeling in biomolecules. 5.2. Modifications in the Spacer Part
- The conversion of compounds 114 and 115 into the hitherto unknown 2’,3’-dideoxy- and2’,3’-dideoxy-2’,3’-didehydro- C-nucleosides 116, 117, 118 and 119 was performed (Scheme 23) [27,
 ,
,  ,
,  ]. These compounds were evaluated in a cell-based assay against Human immunodeficiency 1 (HIV-1). Protection of the 5’-hydroxyl group in 114 and 115 using tert-butyldimethylsilyl chloride and imidazole in anhydrous dimethylformamide or pyridine gave 116 and 117 respectively. The 4-thione product was converted to the 4- methylthio derivative 118 using methyl iodide in aqueous sodium hydroxide. Compounds 116 and 118 were then converted into 1,3-dioxalane-2-thione derivatives 119 and 120 followed by heating in presence of triethylphosphite to yield 121 and 122. Deprotection of these products in presence of tetrabutylammonium fluoride gave the 2’,3’-dideoxy-2’,3’-didehydro-C-nucleosides 123 and 124. Compound 124 reacts with ammonia in methanol at 120ºC under microwave to yield the amino derivative 125. Finally, hydrogenation of 123 and 125 using catalytic amount of Pd/C in ethanol yielded 2’,3’-dideoxy-C-nucleosides 126 and 127.
]. These compounds were evaluated in a cell-based assay against Human immunodeficiency 1 (HIV-1). Protection of the 5’-hydroxyl group in 114 and 115 using tert-butyldimethylsilyl chloride and imidazole in anhydrous dimethylformamide or pyridine gave 116 and 117 respectively. The 4-thione product was converted to the 4- methylthio derivative 118 using methyl iodide in aqueous sodium hydroxide. Compounds 116 and 118 were then converted into 1,3-dioxalane-2-thione derivatives 119 and 120 followed by heating in presence of triethylphosphite to yield 121 and 122. Deprotection of these products in presence of tetrabutylammonium fluoride gave the 2’,3’-dideoxy-2’,3’-didehydro-C-nucleosides 123 and 124. Compound 124 reacts with ammonia in methanol at 120ºC under microwave to yield the amino derivative 125. Finally, hydrogenation of 123 and 125 using catalytic amount of Pd/C in ethanol yielded 2’,3’-dideoxy-C-nucleosides 126 and 127.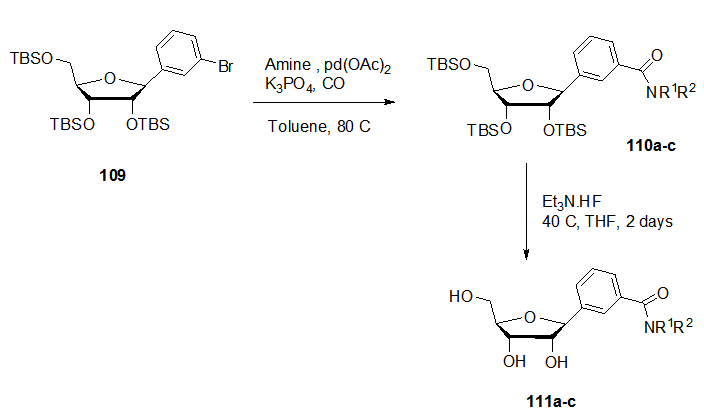 | Scheme 21. Chemical reactions of modification to C-nucleosides 111 |
|
 | Scheme 22. Suzuki-Miyaura cross coupling reaction to prepare C-nucleosides 113 |
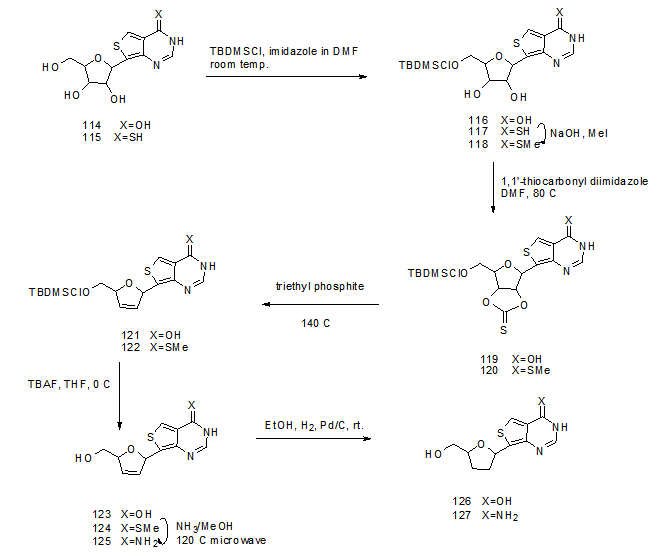 | Scheme 23. Synthetic modification of C-nucleosides 114-115 to 126-127 |
 ], S.-Q. Cao et al synthesized 3’-phosphoramidite derivative 131 starting from 128 (Scheme 24). There 128 was treated with 1.0 M tetrabutylammonium fluoride in THF followed by protecting the 5’-OH group using DMTr to give 129 in good yield. The 2’-OH protected derivative 130 was obtained by action of TBDMSCl on 129. Finally the reaction of 120 with NCCH2CH2OPClN(i-Pr)2 in presence of 2,4,6-collidine and N-methylimidazole gave the phosphoramidite 131.
], S.-Q. Cao et al synthesized 3’-phosphoramidite derivative 131 starting from 128 (Scheme 24). There 128 was treated with 1.0 M tetrabutylammonium fluoride in THF followed by protecting the 5’-OH group using DMTr to give 129 in good yield. The 2’-OH protected derivative 130 was obtained by action of TBDMSCl on 129. Finally the reaction of 120 with NCCH2CH2OPClN(i-Pr)2 in presence of 2,4,6-collidine and N-methylimidazole gave the phosphoramidite 131. 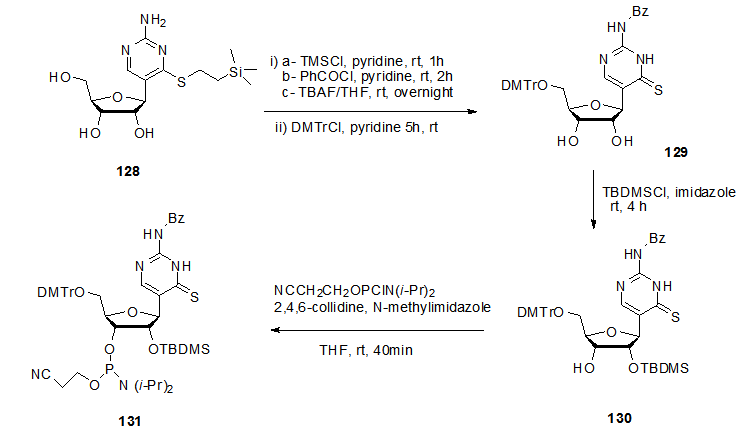 | Scheme 24. Synthetic modification of C-nucleoside 128 to 131 |
5.3. Cyclization of the Acyclic Analogue
- Transformation of the di-O-isopropylidine acyclic C-nucleoside compound 132 to its O-mesyl derivative 133 was performed [29,
 ] by addition of methansulfonyl chloride in pyridine at 0°C for 3 h, followed by deprotection and cyclization to yield the desired cyclic C-nucleoside 134 in good yield by refluxing compound 133 using 4% HCl in 1,2-dimethoxyethane (Scheme 25). The selective cyclization (C-4’-C’-1) and the configuration were confirmed using 13C-NMR and the nuclear overhauser effect (NOE) experiment. Similarly, the isopropylidine derivative 135 was transformed to the cyclic compound 137 under the same conditions through the formation of the tri-O-mesylated derivative 136 (Scheme 26).
] by addition of methansulfonyl chloride in pyridine at 0°C for 3 h, followed by deprotection and cyclization to yield the desired cyclic C-nucleoside 134 in good yield by refluxing compound 133 using 4% HCl in 1,2-dimethoxyethane (Scheme 25). The selective cyclization (C-4’-C’-1) and the configuration were confirmed using 13C-NMR and the nuclear overhauser effect (NOE) experiment. Similarly, the isopropylidine derivative 135 was transformed to the cyclic compound 137 under the same conditions through the formation of the tri-O-mesylated derivative 136 (Scheme 26). 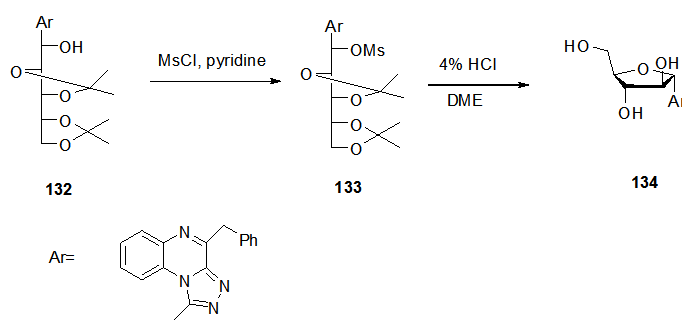 | Scheme 25. Ring closure steps of acyclic C-nucleoside 132 to the cyclic 134 |
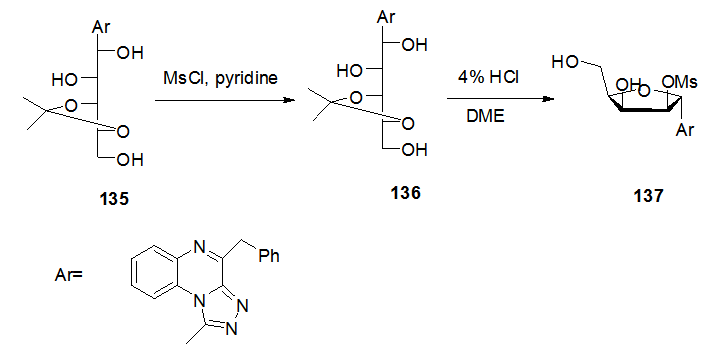 | Scheme 26. Ring closure steps of acyclic C-nucleoside 135 to the cyclic 137 |
 ], where region-selectivity will occur due to the bulkiness of (TIBSCl). This dehydrative cyclization under basic conditions also yields a minor thermodynamically product 140 as a result of the 1,5-SN2 cyclization. During this reaction the intermediate 141 and the byproduct 142 were isolated from the reaction mixture.
], where region-selectivity will occur due to the bulkiness of (TIBSCl). This dehydrative cyclization under basic conditions also yields a minor thermodynamically product 140 as a result of the 1,5-SN2 cyclization. During this reaction the intermediate 141 and the byproduct 142 were isolated from the reaction mixture.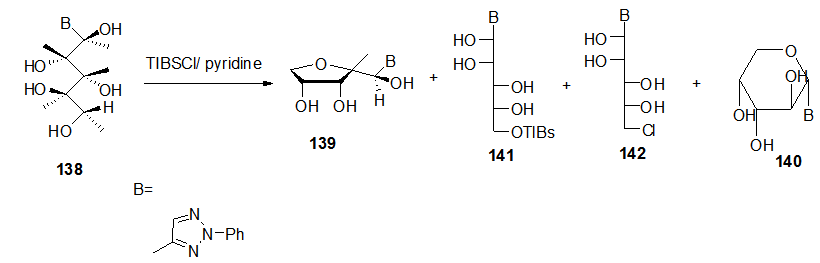 | Scheme 27. Ring closure steps of acyclic C-nucleoside 138 to the cyclic 139-140 |
 ] and the anomeric configuration were determined by the CD and NMR spectroscopic measurements (Scheme 28). Acyclic 5-deoxy-L-manno-pentitol-1-yl nucleoside 143 was treated with methanolic sulfuric acid and subsequent reflux with copper sulphate yield the anomeric mixture of 144 and 145, which were separated by chromatography.
] and the anomeric configuration were determined by the CD and NMR spectroscopic measurements (Scheme 28). Acyclic 5-deoxy-L-manno-pentitol-1-yl nucleoside 143 was treated with methanolic sulfuric acid and subsequent reflux with copper sulphate yield the anomeric mixture of 144 and 145, which were separated by chromatography.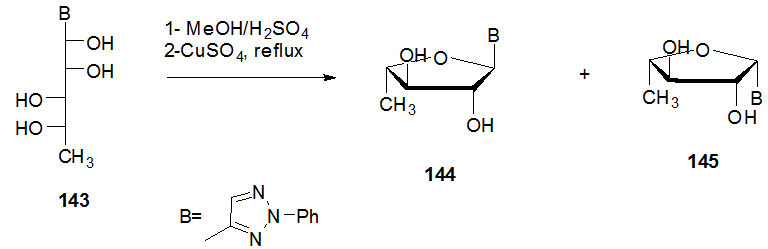 | Scheme 28. Ring closure steps of acyclic C-nucleoside 143 to the cyclic 144-145 |
 ]
] 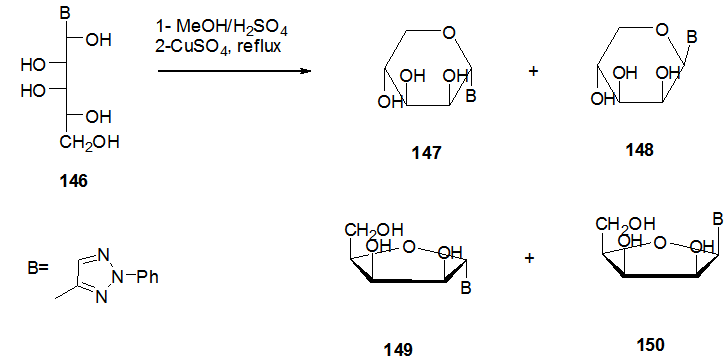 | Scheme 29. Dehydrative cyclization of acyclic C-nucleoside 146 to 147-150 |
6. Miscellaneous Synthesis
- The homo-coupled product 152 was obtained from the reaction of THF-alkyne 151 with different catalytic systems in dry DMF in order to obtain the optimized reaction conditions such as i) CuI, CsCO3 and NaI, ii) CuI and CsCO3 and iii)CuI only, to give the desired product in good yields. It was found that both CsCO3 and NaI have no effect in the reaction (Scheme 30). [33,
 ]The reaction of butenonyl C-glycosides with cyanoacetamide via Micheal addition followed by dehydrative cyclization and oxidative aromatization gave the corresponding glycosyl pyridines. These compounds are evaluated for their antidiabetic potential in vitro. (E)-1-(β-D-glucopyranosyl)-4-(aryl)but-3-en-2-ones 153a-c reacted with cyanoacetamide in DMSO, t-BuOK, N2 and O2 at room temperature followed by acetylation to give the respective 3-cyano-4-(aryl)-6-[(2'',3'',4'',6''-tetra -O-acetyl - β-D-glucopyranosyl)methyl]pyridones 154a-c in good yields. Subsequently, deacetylation of these compounds with NaOMe/MeOH afforded the corresponding 3-cyano-4- (aryl)-6-[(β-D-glucopyranosyl) methyl]pyridones 155a-c (Scheme 31). [34,
]The reaction of butenonyl C-glycosides with cyanoacetamide via Micheal addition followed by dehydrative cyclization and oxidative aromatization gave the corresponding glycosyl pyridines. These compounds are evaluated for their antidiabetic potential in vitro. (E)-1-(β-D-glucopyranosyl)-4-(aryl)but-3-en-2-ones 153a-c reacted with cyanoacetamide in DMSO, t-BuOK, N2 and O2 at room temperature followed by acetylation to give the respective 3-cyano-4-(aryl)-6-[(2'',3'',4'',6''-tetra -O-acetyl - β-D-glucopyranosyl)methyl]pyridones 154a-c in good yields. Subsequently, deacetylation of these compounds with NaOMe/MeOH afforded the corresponding 3-cyano-4- (aryl)-6-[(β-D-glucopyranosyl) methyl]pyridones 155a-c (Scheme 31). [34,  ]
]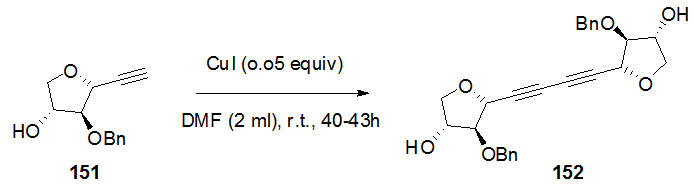 | Scheme 30. C-C coupling reaction of 151 to C-nucleoside 152 |
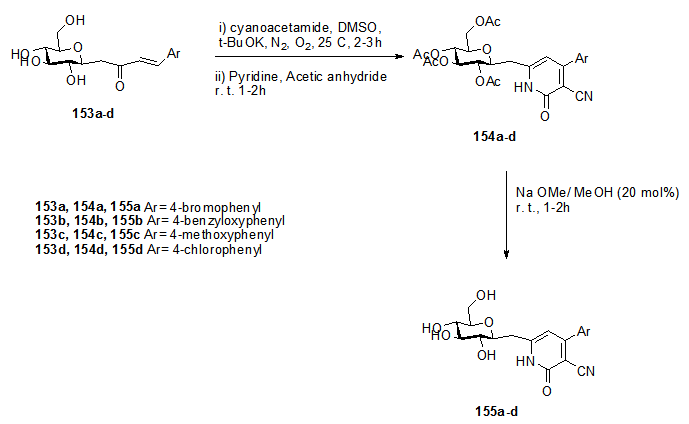 | Scheme 31. Synthetic steps to build up the heterocyclic base on 153-155 |
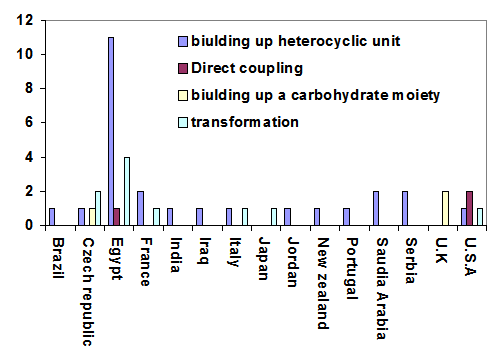 | Figure 2. Distribution of C-nucleosides synthetic tactics by countries |
7. Conclusions
- This work highlights a recent progress in developmental strategies to build up C-nucleosides and analogues. It does not attempt to be comprehensive in particular, it intended to spot locations of work and synthetic tactics (Figure 2), then correlate that to international vs. national transformation of methodologies. From the Middle East, choosing Egypt as a model, it is clear that the internal transformation of research perspectives there dominates. However, science diplomacy may provide a forum for international collaborative research, which leads to intriguing topics of potential applications for sustainable developmentsDedicated to the memory of Prof. Hans W. Zimmer1