-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2012; 2(6): 343-346
doi: 10.5923/j.chemistry.20120206.08
Weak Hydrogen Bonds in Some Six- and Five-atom Interactions: An AIM Topological Analysis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLFrancisco Sánchez-Viesca, Fernando Cortés, Reina Gómez, Martha Berros
Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico. México, D.F., 04510, México
Correspondence to: Francisco Sánchez-Viesca, Faculty of Chemistry, Graduate Division, National Autonomous University of Mexico. México, D.F., 04510, México.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In the 1H NMR spectra of several thiazole derivatives we have found that some of them present downfield shifts (to higher frequency). These ∆∂ can be attributed, in a broad sense, to intramolecular hydrogen bonding. However, we present an AIM study that shows there are two types of atomic interactions in these compounds: some originate bond critical points, but others do not. It is interesting to note that intramolecular hydrogen bonds were formed when a six-member ring and an electronegative atom, such as oxygen or chlorine, were involved. However, interactions involving five-member rings and a nitrogen atom as electron donor showed only three of the six required theoretical properties for hydrogen bonding. These special interactions can be considered as very weak hydrogen bonds. Nevertheless, both types of atomic interactions caused similar downfield shifts in the 1H NMR spectra. Thus, this study reveals differences in the nature of the atomic interactions that gave rise to downfield shifts in the 1H NMR spectra.
Keywords: Intramolecular Hydrogen Bonding, Electron Donors, Atomic-Ring Interactions, Structural Analysis, Physical Chemistry
Cite this paper: Francisco Sánchez-Viesca, Fernando Cortés, Reina Gómez, Martha Berros, "Weak Hydrogen Bonds in Some Six- and Five-atom Interactions: An AIM Topological Analysis", American Journal of Chemistry, Vol. 2 No. 6, 2012, pp. 343-346. doi: 10.5923/j.chemistry.20120206.08.
1. Introduction
- Among the so-called non-conventional hydrogen bonds[1], there is the R-C-H---X group (X=O, Cl and N). These are weak hydrogen bonds[2],[3], since the energy of these secondary bonds is smaller due to the lower acidity of the involved hydrogen atoms.In a previous paper[4], we discussed the 1H NMR spectra of several poly-substituted 4-aryl thiazoles. Our study found two groups of compounds, having or not, the possibility to form C-H---X hydrogen bonds, X being O, Cl or N. The formation or not of these hydrogen bonds gives rise to preferred conformations (rotamers). Figure 1.In this study, three representative compounds were chosen: 2-methyl-4-(2,4,5-trimethoxyphenyl) thiazole, I, which presents downfield shifts in the 1H NMR spectrum (CDCl3, 300 MHz); 2-methyl-4-(2-methyl-4,5-dimethoxyphenyl) thiazole, II, with normal chemical shifts; and 2-methyl-4-(2-chloro-4,5-dimethoxyphenyl) thiazole, III, which presents smaller downfield shifts than compound I. The ∂ values are given in the formulas and we can observe significant ∆∂ for H-6 (benzene ring) and for the thiazolic proton in compounds I and III, as compared to the 2-methylaryl compound II. These downfield shifts (to higher frequency) were ascribed to hydrogen bonding.However, we wanted to confirm, or not, the existence of hydrogen bonding suggested by the experimental NMR chemical shifts results, by a theoretical study of the electronic density of the intramolecular hydrogen bond network of these molecules, as well by H-bond parameters in terms of the bond distances and the appropriate bond angles and compared to other work. The hydrogen bond strengths were calculated by rotating about the C-C bond between the rings, breaking the H bonds. Frequency calculations were done to insure the structures are minima.
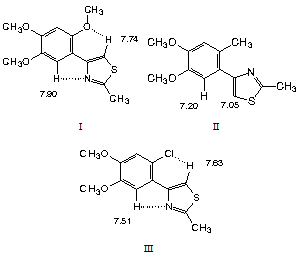 | Figure 1. Hydrogen bonds suggested by 1H NMR study |
2. Computational Details
- Structures of three molecules were optimized at the B3LYP/6-311G++(d,p) level of theory[5-7], with the Gaussian 03 software[8]. Revision C.02 of Gaussian was used to perform the geometry optimizations and produce the wave function files that the AIM analysis needs, AIM 2000 code[9]. The calculations were made in the SGIOrigin 2000/32 supercomputer, at DGSCA-UNAM.
3. Results and Discussion
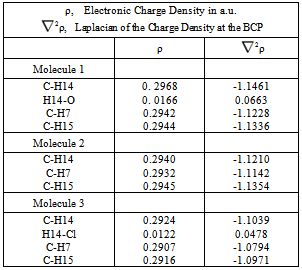
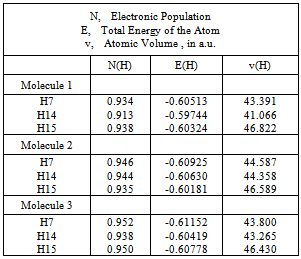
- In order to simplify the calculations, a hydrogen atom replaced the methyl group at the thiazole ring. This change is valid since it does not affect the possibility of hydrogen bond formation. In order to facilitate comparisons, equivalent Arabic numerals are used for these related molecules and the drawing is in accord with the molecular graphs obtained (Figure 2). The structures of molecules 1, 2 and 3 were calculated and a search for bond critical points (BCP) of the C-H---O, C-H---Cl and C-H---N contacts was conducted. In Atoms in Molecules theory, the existence of a bond path between the hydrogen bond donor and the hydrogen bond acceptor, as well as a bond critical point, are sufficient criteria to establish bond existence[10]. The molecular structure can be obtained in a molecular graph showing all the bond paths and bond critical points. The existence of a C-H---O hydrogen bond in the trimethoxyphenyl thiazole 1 is shown in Figure 2. A six member ring is formed and the ring critical point can be observed. The methyldimethoxyphenyl thiazole, 2, presents classical bonding, with no hydrogen bond, as was postulated in[4] for compound II.Molecule 3, the chlorodimethoxyphenyl thiazole, shows a C-H---Cl hydrogen bond, similarly to molecule 1.However, here comes a difference. A possible C-H---N hydrogen bond involving H7 was searched in molecules 1 and 3, but it was not found. With molecule 1, the system was rotated to obtain a zero N-C-C-C-H7 dihedral angle, a planar system, but the N---H7 critical point was not found (although the change was only 0.5°). A similar procedure with biphenyl has been described by Cioslowsky[11]. Therefore, the level of theory was improved to MP2-6-311++G(2d,2p) but the critical point was missing.Nevertheless, the preferred rotamers (1, 2 and 3) are in accord with those previously proposed[4] for compounds I, II and III, and the interaction between the nitrogen atom and H7 has been registered in the 1H NMR spectra of I and III as ∆∂ (downfield shifts).Molecule 3 shows a 26.8° dihedral angle; however, a single-crystal X-Ray diffraction study of the related compound III shows a planar structure[12]. This resembles the biphenyl dihedral angle variations: 26.8° in crystalline state, 19-32° in solution, and 44° in gas phase[13]. Popelier studied extensively C-H---O hydrogen bonds and, based on Atoms in Molecules Quantum Theory (AIM formalism), established a series of charge-density-based criteria for hydrogen bonding identification purposes[14],[15]. His work has been cited many times, C-H---O hydrogen bonds have gained acceptance and have explained conformational stability in some systems[16].The hydrogen bond criteria are:1) The existence of a bond path, containing a bond critical point (BCP) between the donor hydrogen nucleus and the acceptor.2) The second necessary condition involves a local property, namely, the electronic charge density evaluated at the bond critical point. The range of ρ values must be between 0.002 a.u. and 0.035 a.u.3) The value of the Laplacian, Bader[17], of the charge density at the BCP must be between 0.024 a.u. and 0.139 a.u. It is crucial that
 2ρ is positive.4) Loss of charge of the hydrogen atom. This is computed by subtracting the electronic population of the hydrogen in the free state from the associate state.5) Must be energetic destabilization of the hydrogen atom. This effect is measured by ∆E (H).6) Decrease of the hydrogen atom’s volume.In this study, the bond critical point characteristics are summarized for comparison in Table 1. The atomic properties of H7, H14 and H15 in structures 1, 2 and 3 can be found in Table 2. The C-H14---O hydrogen bond satisfies all the above criteria. It presents an adequate topology and the charge density at the H14---O bond critical point is 0.016 a.u. and in molecule 3 the charge density at the H14---Cl BCP is
2ρ is positive.4) Loss of charge of the hydrogen atom. This is computed by subtracting the electronic population of the hydrogen in the free state from the associate state.5) Must be energetic destabilization of the hydrogen atom. This effect is measured by ∆E (H).6) Decrease of the hydrogen atom’s volume.In this study, the bond critical point characteristics are summarized for comparison in Table 1. The atomic properties of H7, H14 and H15 in structures 1, 2 and 3 can be found in Table 2. The C-H14---O hydrogen bond satisfies all the above criteria. It presents an adequate topology and the charge density at the H14---O bond critical point is 0.016 a.u. and in molecule 3 the charge density at the H14---Cl BCP is 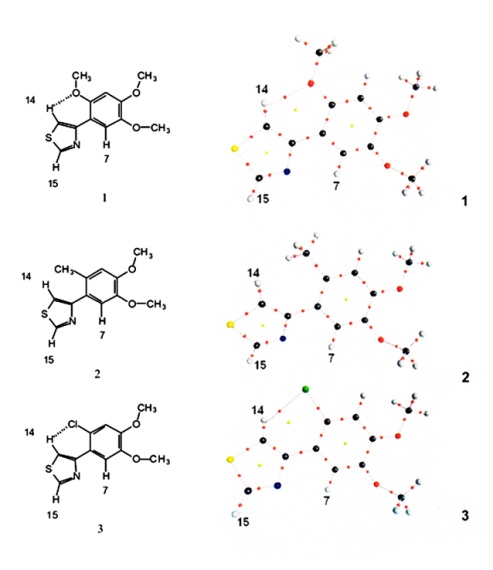 | Figure 2. Molecular graphics of structures 1, 2 and 3, showing the critical points |
|
|
4. Conclusions
- In our compounds 1 and 3, C-H---O and C-H---Cl interactions originate bond critical points and there is agreement with the hydrogen-bond criteria. On the other hand, the C-H---N interactions found by 1H NMR in the same compounds, did not originate bond critical points and are only in partial agreement with hydrogen-bond criteria. These special interactions maybe could be named ‘semibonds’.