-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2012; 2(5): 255-262
doi: 10.5923/j.chemistry.20120205.03
Computational Insights into the Role of the Frontiers Orbital in the Chemistry of Tridentate Ligands
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLF. A. La Porta 1, 2, J. O. S. Giacoppo 2, P. H. Ramos 2, M. C. Guerreiro 2, T. C. Ramalho 2
1Department Physical Chemistry, Paulista State University, CEP 14801-970, Araraquara, SP, Brazil
2Department of Chemistry, Federal University of Lavras, CEP 37200-000, Lavras-MG, Brazil
Correspondence to: F. A. La Porta , Department Physical Chemistry, Paulista State University, CEP 14801-970, Araraquara, SP, Brazil.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The FERMO concept emerges as a powerful and innovative implement to investigate the role of molecular orbitals applied in the description of breakage and formation of chemical bonds. In this work, Hartree-Fock (HF) and Density Functional Theory (DFT) calculations were carried out for a series of four tridentate ligands and their behavior was analyzed using molecular orbital (MO) energies. It was observed that HOMO energies are inadequate to describe the Pearson´s Hard and Solf acid-base (HSAB) pricinple behavior of these compounds. By using the frontier effective-for-reaction molecular orbital (FERMO) concept, the reactions that are driven by HOMO, and those that are not, can be better explained, independent of the calculation method used, since both HF and Kohn-Sham methodologies lead to the same FERMO.
Keywords: FERMO, TridentateLigands, HSAB, HOMO-LUMO, Molecular Orbital
Cite this paper: F. A. La Porta , J. O. S. Giacoppo , P. H. Ramos , M. C. Guerreiro , T. C. Ramalho , "Computational Insights into the Role of the Frontiers Orbital in the Chemistry of Tridentate Ligands", American Journal of Chemistry, Vol. 2 No. 5, 2012, pp. 255-262. doi: 10.5923/j.chemistry.20120205.03.
Article Outline
1. Introduction
- Among the various organic ligands that are used in the synthesis ofcoordination compounds, several studies have been developed with tridentate ligands[1-6].These ligands coordinate to the metal through three atoms and spectroscopic properties are of interest in various areas ofscience such as molecular magnetic materials, lasers in optical fiber and luminescent sensors[7-12]. Molecular orbitals (MOs) and their properties, like energies and symmetries, are very useful for chemists. According to Fukui, the MO properties have used the frontier electron density for predicting the most reactive position in π-electron systems[13-15]. Hoffmann and Woodward set out orbital symmetry rules to explain several types of reactions in conjugated systems, and the frontier MOs gained importance for the better understanding of chemical reactions. The concept of frontier orbital, introduced by Fukui around 1952, relates reactivity with the properties of two molecular orbitals: HOMO and LUMO. One application of the HOMO-LUMO argument is the description of the acid-base behavior of compounds[13-17].Thus, given the limitations of HOMO-LUMO argument and the new approaches proposed in the literature to understand the chemical reactivity, one must move forward the role of molecular orbitals in chemistry, giving rise to a new idea: the Frontier Effective-for-Reaction Molecular Orbital (FERMO)[18-28].The FERMO concept comes from a dose of intuition, together with the criteria for the composition and location to correctly determine the reactant molecular orbital. This concept can be understood as a complement to the HOMO-LUMO argument. Another important feature in FERMO is that both HF and Kohn-Sham orbitals take the same conclusions about reactivity[23-30].In line with those expectations, the goal of the current work is devoted to investigating which MO is the best for describing the reactivity for some systems formed by tridentate ligands, such as the 2-6 pyridinedicarboxylic acid in light of the FERMO concept.
2. Computional Methods
- In recent years, theoretical methods based on the Density Functional Theory (DFT) have emerged as an alternative to traditional ab initio methods in the study of structure and reactivity of chemical systems[31,32]. In this work, all calculations were carried out with the Gaussian03 package[33]. Each tridentate ligands of all 5 compounds was fully optimized using DFT with the B3LYP functional [34,35] employing the 6-31G(d,p) basis set. No symmetry constraint was imposed during the optimization process. Those optimized geometries were used in all subsequent calculations. This theoretical level was also used for the frequency calculations. Furthermore, ab initio MP2 energy calculations were computed using the 6-31G(d,p) basis set in gas-phase. The MO figures were prepared using the Gauss View 2.1 package[33] with a contour value of 0.020. For the calculations, the iron-dipicolinic acid complexes were calculated and characterized by DFT. The energy minima were identified by building potential energy surfaces, obtained along the reaction pathway at the B3LYP/6-31g(d,p) and (8s6p4d1f) level[34]. Furthermore, after each optimization, the nature of the stationary point was established by calculating and diagonalizing the Hessian matrix (force constant matrix). The unique imaginary frequency associated with the transition vector (TV) was characterized. This computational procedure has been used previously in similar systems with success[36-39]. For all calculations of the structures of this work were calculated in gas phase and was not considered the effect of the solvent.
3. Results and Discussion
- In this work, we report the study to show how the FERMO concept can be used to explain the hard and soft behavior for the tridentate ligands (Figure 1). These ligands were chosen because a large number of their experimental and theoretical studies are available in the literature[40-47]. Therefore, these investigations lead to new perspectives and ideas about the reactivity. Reactions involving electron donation and acceptance are related to MO energies, since electrons are occupying and will occupy a MO and a frontier one, as stated by Fukui. By exploration of Pearson’s hard-soft acid-base (HSAB) concept, Klopman proposed the concept of charge or frontier controlled reactions[48,49]. Following the Pearson´s principle that soft molecules have smaller HOMO-LUMO gaps when compared with hard ones, it can be stated that soft reaction sites in a molecule will have a smaller FERMO-LUMO gap than harder ones. Clearly, the HOMO-LUMO gap itself can not describe the hardness difference between the two binding sites in the same molecule. The very chemically intuitive Frontier Effective-for-Reaction Molecular Orbital (FERMO) concept was introduced to solve HOMO-LUMO limitations and better explain the chemical behavior of molecules.
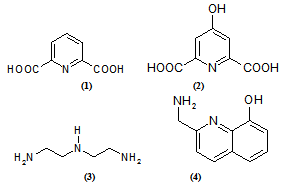 | Figure 1. Structure of the tridentate ligands |
|
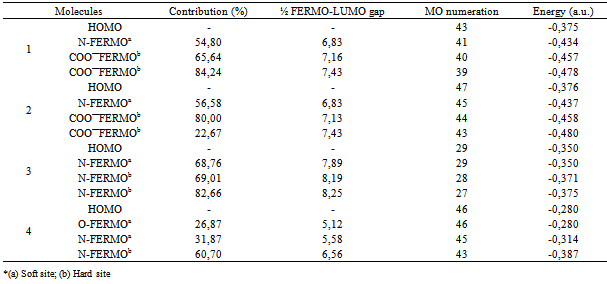
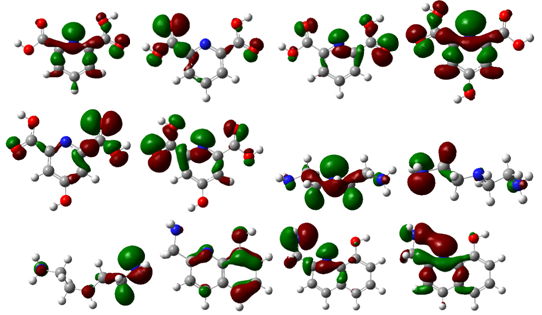 | Figure 2. Surface plots for FERMOs at the MP2 level for the tridentate ligands |
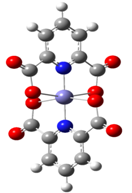 | Figure 3. Model structure Fe-PDC |
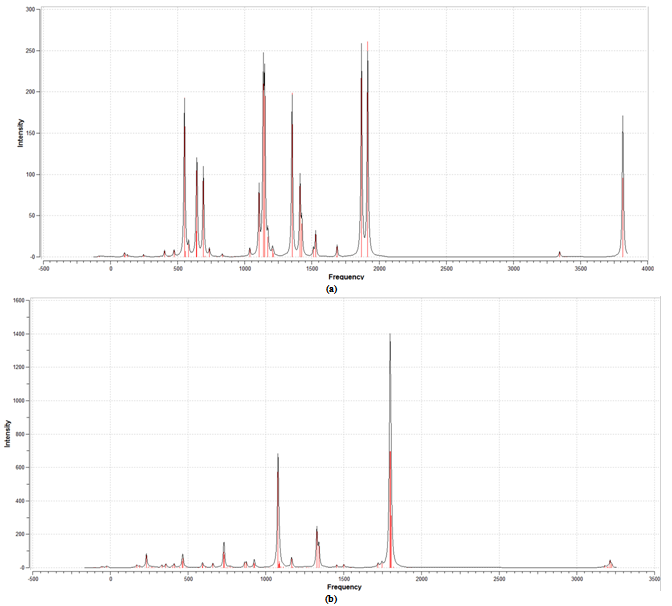 | Figure 4. FTIR spectrum (a) the free ligand and (b) Fe-PDC |
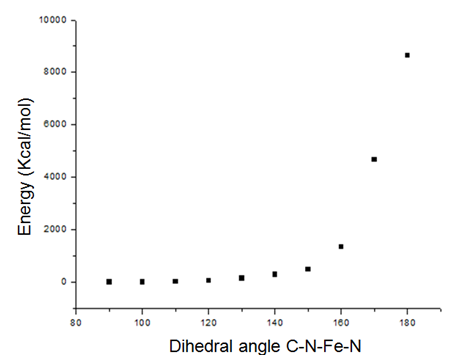 | Figure 5. PES for the Fe-PDC complex structure |
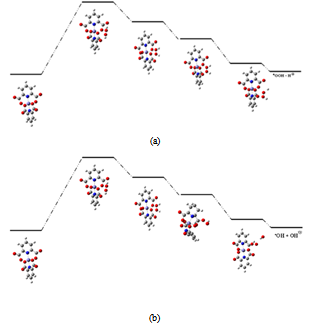 | Figure 6. Catalytic cycle proposed model structures |
4.Conclusions
- Overall, the FERMO concept has been successfully applied to describe to the Pearson´s hard and soft principle in a very simple and chemically intuitive way. The results of this study demonstrated that the complex of iron and acid dipicolínico solid is a good catalyst for the oxidation of quinoline by a Fenton-like mechanism. The results obtained by first principles calculation suggest that proposed mechanism is thermodynamic favorable. Vibrational analysis confirm the formation of the complex Fe-PDC compared with the free ligand spectrum. Thus, the FERMO concept cay be useful to explain acid-base reactions in a wide range of applications. Nevertheless, it is the FERMO concept (the shape of the reactant orbital) that really provided chemical insight about which is the MO related to acid-base reactions. Understanding the behavior of the molecular orbital is crucial to understand the chemistry.
ACKNOWLEDGEMENTS
- We thank the Brazilian agencies CAPES, CNPq and FAPEMIG for funding part of this work. We also are especially grateful to the CEANAPD-SP for the computational facilities.