-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2012; 2(2): 57-82
doi:10.5923/j.chemistry.20120202.11
Electron Transfer Reactions: a Treatise
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLV. Jagannadham
Department of Chemistry, Osmania University, Hyderabad, 500007, India
Correspondence to: V. Jagannadham, Department of Chemistry, Osmania University, Hyderabad, 500007, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In the introduction the concept of oxidation-reduction is well defined. In the discussion a systematic classification of electron transfer reactions is made based on the nature of the redox partners. There were total of eight sets of redox reactions under two main headings four sets for each class of outer sphere electron transfer reactions, and inner sphere electron transfer reactions: containing (i) Inorganic oxidant and inorganic substrate, (ii) Inorganic oxidant and organic substrate, (iii) Organic oxidant and organic substrate, and (iv) Organic oxidant and inorganic substrate. At least one example for each set of redox partners is given and well explained. At the end an example of atom transfer reaction, inner sphere synchronous three and four electron transfer reactions are included.
Keywords: Electron Transfer Reactions, Inner Sphere Electron Transfer Reactions, Outer Sphere Electron Transfer Reactions
Cite this paper: V. Jagannadham, Electron Transfer Reactions: a Treatise, American Journal of Chemistry, Vol. 2 No. 2, 2012, pp. 57-82. doi: 10.5923/j.chemistry.20120202.11.
Article Outline
- 3.3.3.3. Kinetics of Oxidation of Aliphatic Alcohols by Thallium(III): Ruthenium(III) Catalysis [59]
- 3.9.7. Kinetics and Mechanism of in situ Bromohydrination of Cinnamic Acids by N-Bromoacetamide [75]
1. Introduction
- First one must understand the meaning of oxidation and reduction before going to define the concept. The word redox reaction envisages transfer of electron(s), hydrogen atom, hydride ion, oxygen atom, and chlorine atom or chlorinium ion between two redox partners. The word oxidation (of course it necessarily involves concurrent reduction) has been defined in many ways. The most prominent definitions, however, refer to addition of oxygen, removal of hydrogen, or transfer of electron(s), of which the last one is the most favored one at present time. There are several thousands of research publications in literature on different kinds of redox processes. Only some representative publications related to this review were presented and discussed. To understand the nature of the mechanism of a redox reaction it is necessary to find out whether an atom or group or electron transfer has occurred, which atoms are transferred or how many electrons are transferred and what the transition states of all steps are like. For obtaining such information kinetic measurements are useful [1-6]. Acceptance of one electron by an oxidant is equivalent to acceptance of a hydrogen atom and acceptance of two electrons is equivalent to acceptance of a hydride ion. In general most of the redox reactions can be classified into two broad categories following the scheme of Wiberg [7].1. Direct electron transfer reactions.2. Atom, group or ion transfer reactions.
1.1. Direct Electron Transfer Reactions
- Electron transfer reactions may be classified as one equivalent or two equivalent reactions depending on the number electrons being transferred during the oxidation. This kind of classification was first given by Kirk and Brown [1] in 1928. The oxidants like Mn(III), Ce(IV), Fe(III), Cu(III), Ag(II), Co(III), Ni(III) etc act exclusively as one electron abstractors, while Pb(IV), Ru(III), Ag(III), IO4-, Cl2, N-halo compounds, hypohalous acids, peroxo-anions etc are two-equivalent oxidants. Some of the oxidants like Mn(VII), Cr(VI), Pt(IV), Tl(III), V(V) etc act as both one and two equivalent oxidants depending on the conditions employed. In some special cases Cr(VI) and Mn(VII) acted as three and four electron oxidants respectively.It is generally observed that oxidations by two-equivalent oxidants proceed much faster than one-equivalent oxidants, because during two-equivalent oxidations, no high-energy free radicals are formed and there is overall twice the free energy change. For one-equivalent oxidants the rate is affected by the addition of vinyl monomers or mercuric chloride or in the presence of oxygen, but not in the case of two-equivalent oxidations. These two different reaction pathways can also be distinguished from product analysis and stoichiometry of certain reactions. Direct electron transfer is more likely with simple one-electron oxidants like Ce(IV), Co(III) and Fe(III). But with other one or two-electron oxidants either hydrogen atom or hydride ion abstraction takes place. Oxidation of hydrazine or sulfite has been generally used to distinguish between one and two-electron oxidants [8, 9] on the basis of the products formed. One-equivalent oxidants tend to convert sulfite to dithionate and hydrazine to ammonia and nitrogen according to the following reaction mechanisms: (a and b, eqns. 1 - 5)
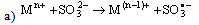 | (1) |
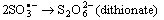 | (2) |
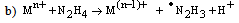 | (3) |
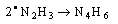 | (4) |
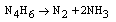 | (5) |
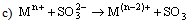 | (6) |
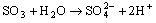 | (7) |
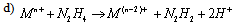 | (8) |
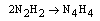 | (9) |
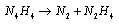 | (10) |
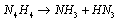 | (11) |
1.1.1. Outer-Sphere Electron Transfer Reactions
- In reactions of this type the metal ion retains its full coordination shell and there is a direct electron transfer from the reductant to the oxidant. The electron given by the reducing agent must be transferred from the primary bond system of one complex to that of the other. The essential feature of this mechanism is that there is no transfer of ligands between the reactants. Kinetically the rate of reaction is faster than the rate of substitution of ligands, or the ligand exchange or displacement is slower than electron transfer. Typical examples of this type are: (eqns. 12 and 13)
 | (12) |
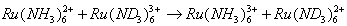 | (13) |
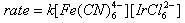 | (14) |
1.1.2. Inner-Sphere Electron Transfer Reactions
- The inner sphere mechanism may be seen, if one of the reactants is labile. In reactions of this class, electron transfers are preceded by the substitution of coordination sphere of one of the ions, with the formation of bridged intermediate in which the two reactants are linked by a common ligand. In this case, ligand displacement is faster than electron transfer process. A typical example of this type is is given in eqn. 15
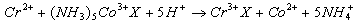 | (15) |
 | (16) |
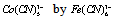 , where both metal ions are substitution inert, a product believed to have a bridged structure. Eqn. 17
, where both metal ions are substitution inert, a product believed to have a bridged structure. Eqn. 17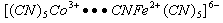 | (17) |
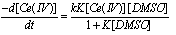 | (18) |
1.2. Atom, Group or Ion Transfer Reactions
- These reactions include1. Hydrogen, oxygen, chlorine atom or group reactions2. Hydrogen atom abstraction reactions.3. Hydride ion abstraction reaction4. Proton abstraction from a precursor-ester or complex.5. Displacement mechanisms.6. Addition-elimination mechanisms.
1.2.1. Hydrogen, Oxygen, Chlorine Atom or Group Transfer Reactions
- When redox reactions take place in aqueous medium the transfer of atom or group is assumed. For example Fe2+ ion may act as a reducing agent by transferring a hydrogen atom from its hydration shell to a substrate eqn.19
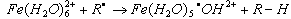 | (19) |
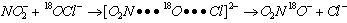 | (20) |
1.2.2. Hydrogen Atom Abstraction Reactions
- In all homolytic cleavages of C-H bonds of organic substrates, abstraction of hydrogen is energetically more favored. Free radical substitution of alkanes (e.g. CH4) with chlorine in the presence of light or heat, one of the propagation steps involves the abstraction of hydrogen, eqn. 21
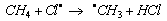 | (21) |
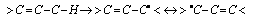 | (22) |
 | Scheme 1. Formation of carbon centered radical |
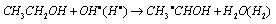 | (23) |
1.2.3. Hydride Ion Abstraction Reaction
- When C-H bond cleaves heterolytically, the hydride ion abstraction can usually occur. Hydride ion abstractions have been proposed in explaining the mechanism of large number of organic reactions such as Meerwein Ponndorff Verley reduction [20], the Cannizaro reaction [21], the Tischenko reaction [22], the Leukart reaction [23], the Sommelet conversion of primary alkyl halides to aldehydes [24, 25], the transition metal ion catalyzed oxidation of organic compounds [26] and many reactions in which a carbocation abstracts a hydride ion intermolecularly or intramolecularly belong to this category [27, 28].As an example the base catalyzed disproportion of aldehydes lacking an α-hydrogen atom (Cannizaro reaction) can be represented as shown in Scheme 2:
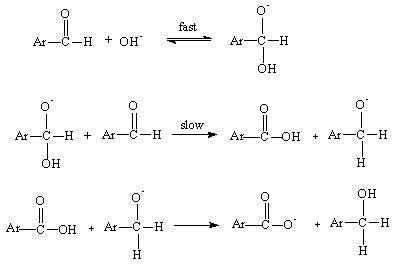 | Scheme 2. Base catalyzed disproportion of aldehyde |
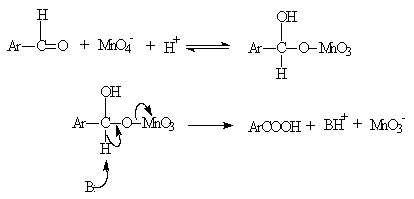 | Scheme 3. Proton abstraction from a precursor-ester or complex |
1.2.4. Proton Abstraction from a Precursor-Ester or Complex
- A number of oxidations are involved the formation of an ester intermediate (usually of an inorganic acid) [34], and then the cleavage of this intermediate by proton loss to any available base. An example of this mechanism can be seen in the oxidation of aromatic aldehydes by permanganate (Scheme 3) [35].The formation of intermediate ester was supported by the presence of 18O in the acid when KMn18O4 was used.The oxidation of glycols by Pb(IV) acetate [36], and periodic acid [37], also followed this pattern, but the positive leaving group is carbon instead of hydrogen. It is seen that whenever proton loss occurs, electron-withdrawing groups accelerate the reaction rate. For instance, Ru(III) catalyzed oxidation of benzaldehydes by IO4- [38], in alkaline medium, where this type of mechanism was proposed, the order of reactivity was 4-NO2 > 3-NO2 > 3-Br > 4-Br > 4-Cl > H > 4-CH3 > 4-OCH3 and the Hammett ρ value was found to be 1.66. However, the observed reverse order of reactivity in acid medium [39] with ρ value 0f - 2.87, points to a hydride ion abstraction mechanism.
1.2.5. Displacement Mechanisms
- In these reactions the organic substrate uses its electrons to cause displacement on an electrophilic oxidizing agent. One example is the addition of bromine to an olefin (Scheme 4) [40].
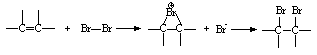 | Scheme 4. Addition of bromine to an olefin |
1.2.6. Addition-Elimination Mechanisms
- In this class of reactions usually peroxy acids or peroxides or their anions involve in the reaction with electron rich organic molecules [40]. The oxidizing agent adds to the substrate and then part of it is lost Scheme 5:
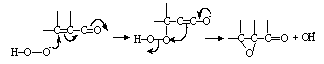 | Scheme 5. Oxidation of olefins by peroxides |
2. Scope of the Review
- The chemical mechanisms of several oxidation-reduction reactions have been well studied and are continued to be studied. The investigations on the kinetics and mechanism of these reactions have played an important role in the development of Physical Organic Chemistry.A mechanism is the actual process by which a reaction takes place - which bonds are broken, in what order, how many steps are involved, the relative rate of each step, etc. In order to state a mechanism completely: one should have to specify the positions of all atoms, including those in solvent molecules, and the energy of the system, at every point in the process. A proposed mechanism must explain all the experimental facts available. It is always subject to change as new experimental facts are discovered. The usual course is that the gross features of a mechanism are the first to be known and then increasing attention is paid to finer details. The tendency is always to probe more deeply, to get more detailed descriptions. Although for most reactions gross mechanisms can be written today with a good degree of assurance, no mechanism is known completely. There is much about the fine details, which is still puzzling, and for some reactions even the gross mechanism is not yet clear. The problems involved are difficult because there are so many variables. Many examples are known where reactions proceed by different mechanisms under different conditions. In some cases there are several proposed mechanisms, each of which completely explains all the data.The original observation that the results of studies on several redox reactions lend themselves to simple generalizations was satisfying and provided confidence that the mechanisms for more complex reactions might also be understood. However, the exceptions to these generalizations show that it is dangerous to assume that even simple reaction mechanisms can be easily understood. These exceptions have the potential to provide new insight into the mechanism for several redox reactions by carrying new experiments. As said in the above paragraphs, a good Physical Organic Chemist is always eager in finding the way to reach a specific goal for the problem that he has in his mind. In this course of time, this review would help for writing gross mechanisms of several synthetically hitherto known important reactions and paves the way to get an insight into finer details.A criticism which is anticipated from the readers of this review is about the inclusion of the first two sections of discussion on inner and outer-sphere electron transfer reactions between inorganic redox partners. One may ask where the organic chemistry or physical-organic chemistry here is. This criticism may spring from different points of view where the lines should be drawn which divide and bound the review. Some topics like these first two sections are treated as essential core material in this review. The title itself of the review warrants inclusion of such topics, because they involve all the essential features of kinetics like stoichiometry of the reaction, and effect of [H+], ionic strength, dielectric constant, temperature, [catalyst] and [complexing agent] on rates. They will be very useful to start with for a beginning kineticist.
3. Discussion
3.1. Outer-Sphere Electron Transfer Reaction between an Inorganic Oxidant and Inorganic Substrates
- Establishment of the nature of the reactive species of the Ni(III) ion - Kinetics of oxidation of iodide and thiocyanate ions by nickel oxyhydroxide in aqueous dilute sulfuric acid medium [42]:Solid NiO(OH) was prepared by the oxidation of Ni(II) ions by bromine in aqueous alkali. The reaction of this NiO(OH) with iodide and thiocyanate ions in aqueous H2SO4 medium was overall second order, being first order each in [oxidant] and [reductant]. The rate law at constant [H+] was found to be as given in eqn. 24
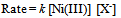 | (24) |
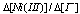 was found to be 1 in the range 0.25 to 1.00 M of H+. It was therefore concluded that the overall reaction could be as follows: eqn. 25
was found to be 1 in the range 0.25 to 1.00 M of H+. It was therefore concluded that the overall reaction could be as follows: eqn. 25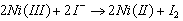 | (25) |
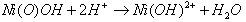 | (26) |
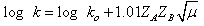 | (27) |
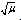 it was concluded that Ni(OH)2+ was assumed to be the reactive species of Ni(III), being iodide to be as I-. A reaction between two species that proceeds via a polar transition state should be sensitive to variation of solvent polarity. If the reaction was of the same sequence as shown below (eqn. 28) should have a solvent effect on rates.
it was concluded that Ni(OH)2+ was assumed to be the reactive species of Ni(III), being iodide to be as I-. A reaction between two species that proceeds via a polar transition state should be sensitive to variation of solvent polarity. If the reaction was of the same sequence as shown below (eqn. 28) should have a solvent effect on rates.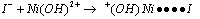 | (28) |
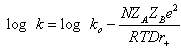 | (29) |
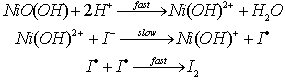 | Scheme 6. Reaction between NI(III) and iodide ion |
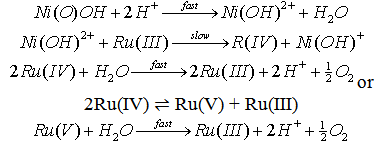 | Scheme 7. Self decomposition Ni(III) ion in presence of Ru(III) |
3.2. Inner-Sphere Electron Transfer Reaction between Inorganic Oxidant and Inorganic Substrates
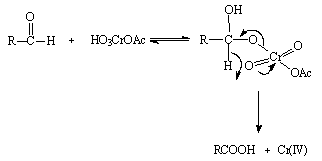
- One-electron reduction of Ni(III) by hydroxylamine and hydrazine via addition/elimination - an example of an inorganic inner sphere electron transfer reaction [44]:The rates of oxidations in aqueous solution of NH2OH and N2H4 by Ni(III) ion were measured iodometrically in the presence and absence of 2,2'-bipyridine (BP). The products of oxidation were N2O and H2O from NH2OH and ammonia and nitrogen from hydrazine respectively. The reaction followed the rate law: eqn. 30
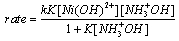 | (30) |
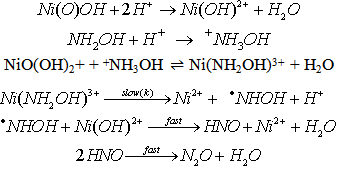 | Scheme 8. Reaction between NH2OH and NI(III) |
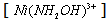 came from the linear plot of 1/ k vs. 1/ [+NH3OH] with a positive intercept on y-axis.
came from the linear plot of 1/ k vs. 1/ [+NH3OH] with a positive intercept on y-axis.3.3. Outer-Sphere Electron Transfer Reaction between an Inorganic Oxidant and Organic Substrates
3.3.1. Chromium(VI) ion
- Cr(VI) oxidation of benzaldehydes [45], benzaldoximes [46], aliphatic esters [47,48], toluenes [49], anisoles [50,51], dimethyl sulfoxide [52] and phenols [53] was carried out in various media. All these reactions obeyed total second order kinetics. Identification of products, discussion of mechanism and application of Hammett and Taft equations were some of the salient features of these reactions.
3.3.1.1. Benzaldehydes [45]
- A search for resonance effects in the kinetics of chromic acid oxidation of a series of benzaldehydes was made: The kinetics of oxidation of a series of eight meta and para-substituted benzaldehydes by chromic acid has been re-investigated in acetic acid water mixture with a view on the search for resonance effects of para-substituents. The overall order of the reaction was two, first order in each reactant at constant acidity. The products of oxidation were the corresponding carboxylic acids. Hammett's reaction constants (ρm) and (ρp) for meta and para-substituents were calculated separately as 0.851 and 1.23 respectively. The reason for the higher ρp value than ρm was due to the direct resonance interaction of para-substituents with the reaction center. The individual contributions of inductive and resonance effects were calculated for para-substituents. The para-substituents did not fit in to the correlation of meta-susbstituents, instead they correlated well with. Based on these results, we proposed a mechanism as shown in scheme 9, which shows a heterolytic cleavage of C-H bond, and which is different from that proposed by Wiberg and Szeimies assuming the homolytic cleavage of aldhydic C-H bond for which one could expect a very low ρ value:
 | Scheme 9. Reaction between aldehyde and Cr(VI) |
 | Scheme 10. Reaction between benzaldoxime and Cr(VI) |
3.3.1.2. Benzaldoximes [46]
- Kinetics of chromic acid oxidation of benzaldoximes in acetic acid-water mixtures: Kinetics of oxidation of a series of six benzaldoximes by chromic acid in 30% (vol./vol.) acetic acid-water mixture was measured at constant acidity and ionic strength. The reaction was over-all second order, being first-order in each reactant. The rates of oxidation were not affected by the addition of salts like Na2SO4 and NaHSO4. The reaction was acid catalyzed with an order in [H+] to be four and the rate of oxidation increased with increase in percentage of acetic acid. Hammett's equation was applicable with a reaction constant ρ = -2.00 and a correlation coefficient r = 0.998 indicating an electron deficient center in the transition state. The Arrhenius plots for four benzaldoximes met at 325 K, which was well above the experimental temperature range indicating the reaction series was considered to be enthalpy-controlled process. Formation of a very unstable cyclic chromate ester in a fast step and subsequent decomposition of this in a slow step had been suggested as a probable mechanism as shown in scheme 10:
3.3.1.3. Aliphatic Esters [47]
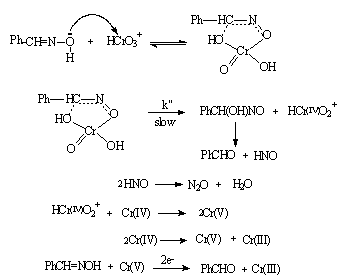
- Kinetics and mechanism of oxidation of some aliphatic esters by chromic acid in the presence and absence of oxalic acid in acetic acid-water medium: The oxidation kinetics of aliphatic esters by chromic acid was examined in aqueous acetic acid in the presence and absence of oxalic acid. In the absence of oxalic acid the reaction was overall second order, being first order in each reactant. It was acid catalyzed and the reaction rates were independent of added salts and it was dependent upon solvent polarity. In the presence of oxalic acid the rate of oxidation increased appreciably. The products of oxidation in the absence of oxalic acid were acetic acid and the corresponding aldehydes (Me2CO in the case of Me2CHOAc). The formation of free radicals was observed only in the presence of oxalic acid. There could be a possibility of ester hydrolysis before the oxidation could take place. The kinetic study was done in 20% aqueous acetic acid in presence of 3.0 M sulfuric acid. Under these conditions the observed rate constant for the hydrolysis of methyl acetate was found to be 7.88 X 10-4 min-1. Since this was much lower than the observed rate of oxidation of methyl acetate by Cr(VI) (1.0 X 10-2 min-1), a direct oxidation of the ester had been envisaged. Activation parameters were calculated and from the isokinetic plot it was concluded that the reaction sequence to be entropy controlled one. A suitable mechanism suggested in the absence of oxalic acid (in the presence of oxalic acid will be discussed later under the heading of direct three electron transfer reactions) with the formation of an initial very unstable complex in the fast step which later gives acetic acid and the corresponding carbonyl compound as the products (scheme 11). The other steps were essentially similar to those mentioned above.
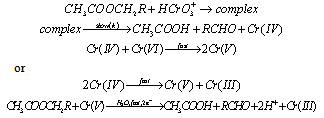 | Scheme 11. Reaction between ester and Cr(VI) |
3.3.1.4. Application of Taft Equation [48]
- Taft four parameter equation as a mechanistic tool in chromic acid oxidation of aliphatic esters: The Taft four parameter equation was used to correlate the kinetic data in the chromic acid oxidation of CH3COOCHRR1 (R = H, Me; R1 = H, Me, Et, Me2CH, Pr). Both polar and steric effects were operative in this reaction. The mechanism involved attack of a positively charged Cr species at the C-H bond of CHRR1. The Taft reaction constants ρ* and δ were found to be -7.14 and 2.72 respectively indicating the electron withdrawing and more bulky groups favoring the reaction.
3.3.1.5. Applicability of the Taft four parameter equation [49]
- Kinetic study of chromic acid oxidation of eight o-substituted toluenes in acetic acid-water mixtures: The kinetics of the Cr(VI) oxidation of o-RC6H4Me (R = MeO, EtO, Me, F, Cl, Br, I, NO2), to the corresponding o-RC6H4CHO in 80% aqueous acetic acid and 1 M HClO4 had poor linear free energy correlation with either ρ* or Es. A four parameter Taft linear free energy relationship, log k/ko = ρ*σ* + δEs was found to be applicable satisfactorily. The polar (P) and steric (S) effects were quantitatively separated. And the polar and steric interaction energies were evaluated by the use of the equations P(polar) = - 2.303 ρ*σ* RT and S(steric) = - 2.303 δEs RT. The total polar and steric interaction energies (P + S) were well correlated with the ΔΔG≠.
 | Scheme 12. Reaction between anisole and Cr(VI) |
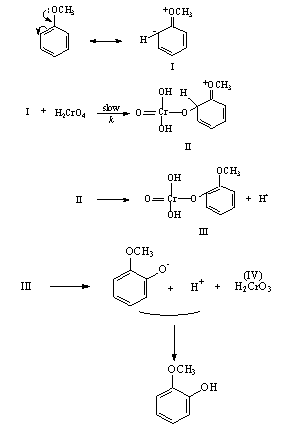
3.3.1.6. 1,10-Phenanthroline Catalyzed and Un-Catalyzed Oxidation of Anisole by Chromic Acid In Acetic Acid-Water Mixture [50]
- The hydroxylation of anisole, to give o - and p - HOC6H4OMe, by H2CrO4 in aqueous acetic acid-H2SO4 was accelerated by the presence of 1,10-phenanthroline as catalyst. The rate-limiting step involved the attack of chromium-phenanthroline complex on anisole. Under otherwise similar conditions there was no reaction between 1,10-phenanthroline and Cr(VI). The mechanism in the absence of 1,10-phenanthroline is shown in scheme 12
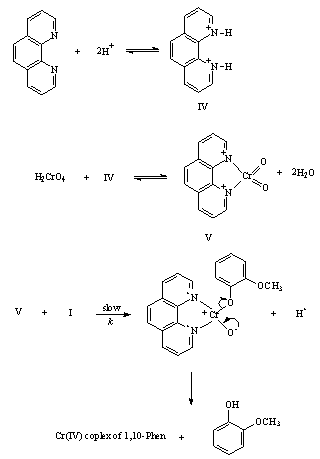 | Scheme 13. The mechanism in presence of 1,10-phenanthroline |
 | Scheme 14. Reaction between anisole and Cr(VI) |
3.3.1.7. Application of Linear Free Energy Relationships (LFER) in the Aromatic Hydroxylation of Anisoles by Chromic Acid in Acetic Acid-Water Mixtures [51]
- An LFER for the rates of the title reaction, which was first order in each of substrate and oxidizing agent at constant H+ concentration and ionic strength, showed that the site of attack was the C atom ortho to the MeO group. The reaction mechanism was suggested as shown in scheme 14:
3.3.1.8. Kinetics and Mechanism of Oxidation of Dimethyl Sulfoxide by Chromic Acid in Aqueous Acid Medium [52]
- The title reaction showed first order dependence each on [oxidant] and [substrate]. The reaction was acid-catalyzed and showed fourth order dependence in [H+]. The rate was affected to a small extent by added sulfate ions and was dependent upon solvent polarity. Added Mn(II) retards the rate while the rate was enhanced if 1,10-phenanthroline was also present along with Mn(II). The rate retarding effect of Mn(II) is expected if Mn(II) ions catalyze the disproportion of the intermediate valency states of Cr(VI) i.e. Cr(IV) or Cr(V). However added 1,10-phenanthroline in the presence of Mn(II) ions catalyzed the rate of oxidation. This might be due to the fact that the added Mn(II) is removed as its complex and the disproportion of the Cr(IV) or Cr(V) was not diminished hence the higher rate of oxidation. This was an additional evidence in Cr(VI) oxidations to show the formation of Cr(IV) and Cr(V) as intermediates. The sole reaction product was identified as dimethyl sulfone. Based on the above discussion, the mechanism could be written as shown in scheme 15 with an initial formation of the complex in a fast step and subsequent decomposition of the complex in the rate-determining step. The remaining steps being similar to those as mentioned in the section 3.3.1.2 above for benzaldoximes.
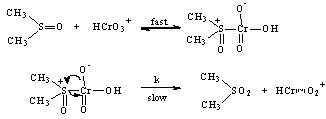 | Scheme 15. Reaction between DMSO and Cr(VI) |
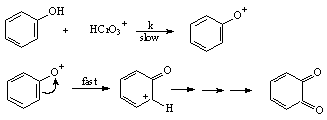 | Scheme 16. Reaction between phenol and Cr(VI) |
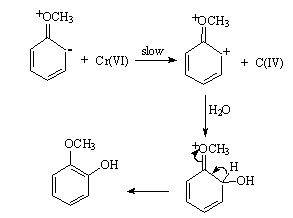
3.3.1.9. Kinetics and Mechanism of Oxidation of Phenols by Chromic Acid in acetic Acid-Water Mixture [53]
- The title reactions were first order each in [chromic acid] and [phenol]. The phenols having electron-donating substituents in the benzene ring accelerated the rates and vice versa indicating an electron deficient center with a Hammett ρ+ of – 3.36. A plot of ΔH≠ vs. ΔS≠ was linear and suggests that the route of the oxidation was same for all the phenols. The effect of H+ and dielectric constant on rates revealed that the reactive species of Cr(VI) to be HCrO3+. A mechanism involving a direct attack of HCrO3+ on the phenolic oxygen atom in the rate-determining step was proposed as shown in scheme 16.
3.3.2. Nickel(III) ion
- Reactions of Ni(III) ion with aliphatic primary [54] and secondary alcohols [55] and glycols [56] were studied. The total order of the reaction was found to be two. The oxidation process of primary and secondary alcohols went through the abstraction of a α-hydrogen. A synchronous carbon-carbon and oxygen-hydrogen bond cleavage was shown to take place in the case of glycols based on the kinetic and entropic evidences. The products of oxidation were the corresponding carbonyl compounds.
3.3.2.1. One-electron Reduction of Ni(III) Ion by Aliphatic Primary Alcohols in Aqueous H2SO4 Medium: A Kinetic and Mechanistic Approach [54]
- The rates of reactions of a series of seven aliphatic alcohols in aqueous solution with Ni(III) ion were studied iodometrically at constant acidity and ionic strength under pseudo-first-order conditions. The overall order of the reaction was two: first order each in [oxidant] and [reductant]. The rate of reduction of Ni(III) was not affected by added salts like (NH4)2SO4 and K2SO4. The reaction was acid catalyzed at constant ionic strength with an order of two in [H+]. The rate of reduction also increased with increase in percentage of acetic acid. The plot of log k vs. 1/D gave a straight line with a positive slope indicating that one of the reactants is a positive ion i.e. Ni(OH)2+. Taft equation was applicable with a reaction constant of ρ* = -1.41 and correlation coefficient r = 0.91). A mechanism involving an outer-sphere electron transfer from alcohol to Ni(III) ion was proposed in the rate determining step which was in consistent with observed kinetic results. Formation of free radicals as reactive intermediates was identified by a positive polymerization of acryl amide. The absence of any reaction between t-butyl alcohol and Ni(III) was a positive indication of a α-C-H bond breakage in the rate determining step. The reduction of Ni(III) ion was characterized by activation enthalpies (53 to 76 kJ mol-1) and strongly negative (-22 to -96 J K-1) activation entropies which originate from hydration of a proton produced in the rate determining step.
3.3.2.2. One-Electron Oxidation of Aliphatic Secondary Alcohols by Ni(III) Ion in Aqueous Sulfuric Acid-Acetic Acid Medium: a Kinetic and Mechanistic Study [55]
- Oxidation of a series of aliphatic secondary alcohols MeCHROH (R = Me, Et, Pr, Bu, hexyl) by Ni(III) in aqueous acetic acid and H2SO4 medium at constant acidity and ionic strength exhibited overall second-order kinetics. The rates increased with increasing [H+] at constant ionic strength. The Taft equation gave ρ* = -1.00 at 303 K. A mechanism involving an outer sphere electron transfer from alcohol to Ni(III) ion was proposed. The mechanism of oxidation of both primary and secondary alcohols can be depicted as shown in scheme 17.
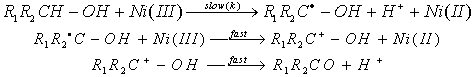 | Scheme 17. Reaction alcohols with Ni(III) |
3.3.2.3. A Synchronous C-C and O-H Bond Cleavage of Some 1,2-Diols by Ni(III) Ion in Sulfuric Acid Mdium: a Kinetic, Entropic and Mechanistic Aproach [56]
- The kinetics of oxidation of HOCHRCHR1OH (R = R1 = H, Me; R = H, R1 = Me, Et) by Ni(III) ion was examined in aqueous H2SO4. The rates were affected by acidity and dielectric constant but not by added SO42- ions. The reaction was overall second order, first order in each reaction component, and proceeded via a synchronous C-C and O-H bond cleavage, which was confirmed by product analysis, kinetic parameters and large negative entropies of activation. Ni(OH)2+ was shown to be the reactive species from the application of Amis equation to the rate data at different solvent compositions. From the product analysis, stoichiometry, presence of free radicals and foregoing kinetic results the mechanism was given as shown in scheme 18 taking ethylene glycol as a typical example:
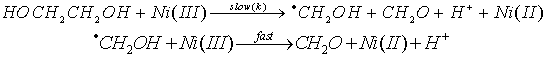 | Scheme 18. Reaction between glycol and Ni(III) |
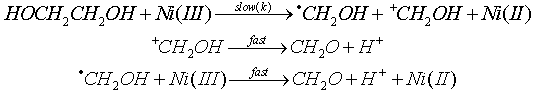 | Scheme 19 |
3.3.3. Thallium(III) ion
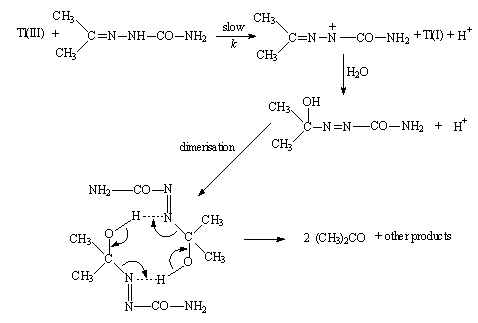
- Oxidation of acetophenone-semicarbazones, acetone –semicarbazones [57], the ruthenium(III) adducts of ethanolamines [58], aliphatic alcohols [59] and mannitol [60] were studied with Tl(III) ion. All these reactions followed total second order kinetics. The pathway of semicarbazones took place via a dimer of the hydroxy semicarbazone that was first formed in the rate-determining step by a α-hydrogen abstraction followed by reaction with water. There was no reaction of Tl(III) ion with ethanolamines, alcohols and mannitol in the absence of Ru(III). Here Ru(III) was used as a catalyst.
3.3.3.1. Kinetics of Oxidation of Semicarbazones by Thallium(III) [57]
- Oxidation of semicarbazones of acetone, methyl ethyl ketone, acetophenone and 4'-methylacetophenone by Tl (III) in aqueous acetic acid containing H2SO4 and Cl- was examined. The reaction followed a total second order. The rate of oxidation decreased with increasing [H+] and with increasing [Cl-]. These two observations could be interpreted with the following (eqns. 31 and 32) two equilibriums assuming Tl(OAc)2+ as the reactive species.
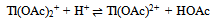 | (31) |
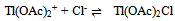 | (32) |
 | Scheme 20. Reaction of semicarbozones with Tl(III) |
3.3.3.2. Kinetics of Oxidation of Ethanolamines by Thallium(III): Ruthenium(III) Catalysis [58]
- Kinetics of oxidation of monoethanolamine, diethanolamine and triethanolamine by Tl(III) with Ru(III) as catalyst has been studied in acetic acid-water mixture. The reaction was first order in [Tl(III)] and fractional order in [substrate] and [Ru(III)]. The reaction ceased even in lower concentrations of H2SO4 and NaCl. The rate decreased with increase in percentage of acetic acid. This revealed that the reactive species of thallium in aqueous acetic acid was supposed to be neutral Tl(OAc)3. A mechanism involving formation of an adduct between the catalyst and the substrate in a fast step which reacts later with the oxidant in a slow step to give products has been proposed. Thermodynamic and activation parameters for formation of the adduct and for its reaction with Tl(III) have been determined respectively. The negative ΔS values may be attributed to the more rigid nature of the adduct formed, which also accounted for the decrease in number of molecules on going from the reactants to the adduct formation stage. The negative ΔS≠ values were in agreement with formation of an H+ in the rate determining step, which could be solvated hence freezing the movement of free molecules. The non-involvement of the free radical intermediates was confirmed by negative polymerization test. The products of the reaction were found to be HCHO and NH3.
3.3.3.3. Kinetics of Oxidation of Aliphatic Alcohols by Thallium(III): Ruthenium(III) Catalysis [59]
- Kinetics of oxidation of aliphatic alcohols by Tl(III) with Ru(III) as catalyst were studied in acetic acid. The reaction was first order in [Tl(III)] and fractional order in [substrate] and [Ru(III)]. The reaction ceased even in low concentrations of H2SO4 and NaCl. The rate decreased with increasing percentage of acetic acid, indicating [Tl(OAc)3] was the reactive species. The noninvolvement of free radical intermediates was confirmed by a negative polymerization test. Mechanisms involving adduct formation between the catalyst and substrate in a fast step and the oxidation of this adduct by the oxidant in the rate determining step to form the products had been proposed. The product of the reaction was the corresponding carbonyl compound.
3.3.3.4. Kinetics and Mechanism of Oxidation of Mannitol by Thallium(III): Ruthenium(III) Catalysis [60]
- The rates of oxidation of D-mannitol by Tl(III) with Ru(III) as catalyst had been measured in acetic acid medium. The reaction was first order in [Tl(III)], fractional order each in [substrate] and [Ru(III)]. The reaction ceased even in lower concentrations of H2SO4 and NaCl. The rate decreased with increase in percentage of acetic acid. This revealed that the reactive species of thallium in aqueous acetic acid was supposed to be Tl(OAc)3. The non-involvement of the free radical intermediates was confirmed by negative polymerization test. Mechanisms involving adduct formation between the catalyst and substrate in a fast step and the oxidation of this adduct by the oxidant in the rate determining step to form the products had been proposed. The product of the reaction was D-mannose.The rate law (eqn. 33) and the mechanism (scheme 21) which could explain all the studies of above three redox processes are given below:The rate law:
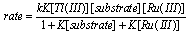 | (33) |
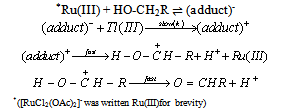 | Scheme 21 |
3.4. Inner-Sphere Electron Transfer Reaction between an Inorganic Oxidant and Organic Substrates
3.4.1. Cerium(IV) ion: Silver(I) Catalyzed and Un-catalyzed Oxidation of Dimethyl Sulfoxide by Ceric Nitrate in Nitric Acid Medium [61]
- The un-catalyzed oxidation of DMSO by Ce(IV) involves complex formation in HNO3 medium which yields dimethyl sulfone as the final product. The evidence for the complex formation between Cr(IV) and DMSO was obtained from kinetic observations and the absorption spectra of both Ce(IV) and Ce(IV)-DMSO complex which showed a shift in λmax from 340 nm to 360 nm. The orders in [Ce(IV)] and [DMSO] were one and fractional respectively. The intra molecular electron transfer in the complex resulted in the liberation of DMSO+• radicals which in subsequent steps convert in to sulfone. The formation of free radicals was confirmed by acrylonitrile polymerization. With Ag(I) as catalyst, an Ag(I)-DMSO adduct was proposed. This adduct reacts with Ce(IV) bimolecularly to give a Ag(II)-DMSO adduct which yields the products. The evidence for the formation of Ag(II) as the intermediate was obtained from kinetic features and spectral studies. When bipyridyl added to the reaction mixture a brown colored complex of Ag(II) with a characteristic maximum at 454 nm was obtained supporting the formation of Ag(II)-DMSO adduct as the intermediate in the catalyzed process. The kinetic evidence for the formation of intermediate complexes both in the catalyzed and un-catalyzed processes was the linear double reciprocal plot obtained when 1/kobsd was plotted against 1/[substrate] or 1/[catalyst]. The rates exhibited fractional order in [Ag(I)] as well as [DMSO] and first order in [Ce(IV)]. The other kinetic features observed in the catalyzed process were same as that observed in the absence of Ag(I). From the effect of [H+] on rates it was concluded that the neutral species of cerium i.e. Ce(NO3)4 was the reactive one in the catalyzed and un-catalyzed reactions. In the catalyzed redox process, there could be a possibility of competition between Ag(I) and Ce(IV) for DMSO to give Ag(I)-DMSO adduct or Ce(IV)-DMSO complex. Analysis of the kinetic data from double reciprocal plots the formation constants K (in the absence of catalyst) and Kcat (in the presence of catalyst) were found to be 9.0 mol-1 and 30.3 mol-1. The value of 9.0 mol-1 for K (the formation constant of Ce(IV)-DMSO complex) in the absence of catalyst is far less than the value of 30.3 mol-1 for Kcat (the formation constant of Ag(I)-DMSO adduct) in the presence of catalyst. This clearly indicated that the Ag(I)-DMSO adduct was exclusively formed in the presence of Ag(I). Based on these results the mechanisms suggested were as follows (Schemes 22 and 23):Uncatalyzed:
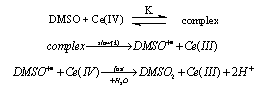 | Scheme 22. Reaction of DMSO with Ce(IV) |
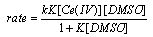 | (34) |
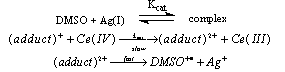 | Scheme 23. Reaction of DMSO with Ce(IV) in presence of Ag(I) |
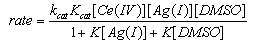 | (35) |
3.4.2. Nickel(III) ion
- Oxidation of benzaldehydes [62], α-hydroxy acids [63] and ethanolamines [64] has been studied. In all the three systems studied, the rate law was found to be as shown in eqn. 36
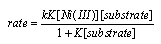 | (36) |
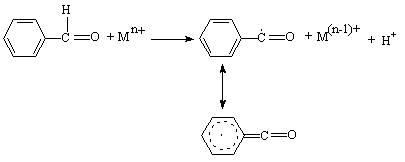
3.4.2.1. Kinetics and Mechanism of Oxidation of Benzaldehydes by Nickel(III) Ion in Acetic Acid-Water Mixtures [62]
- The rates of oxidation of a series of benzaldehydes (BA) by Ni(III) ion were measured in acetic acid-water mixtures at constant acidity [H2SO4] and ionic strength. The reaction was fractional order in [BA] and first order in [Ni(III)]. The reaction was acid catalyzed, with an order of two in [H+], and was unaffected by added Na2SO4. Also the rates were sensitive to solvent polarity. Hammett's equation was applicable with a reaction constant ρ = 1.92 and correlation coefficient r = 0.978. Formation of an intermediate in a fast step and subsequent decomposition of this in a slow step by a homolytic cleavage of aldehydic C-H bond was proposed as a probable mechanism. The presence of free radical intermediates was confirmed by acryl amide polymerization test. From the iso-kinetic plot the reaction series was considered to be entropy controlled one. The product was identified as benzoic acid from its melting point. The mechanism was as given in scheme 24.
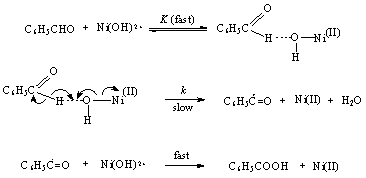 | Scheme 24. Reaction between benzaldehydes and Ni(III) |
3.4.2.2. Oxidation of Lactic Acid (LA) and Mandelic Acid (MA) by Nickel(III) Ion in Sulfuric Acid Medium Via Addition/Elimination [63]
- The oxidation of lactic acid and mandelic acid by Ni(III) ion was studied in aqueous acid medium. A rapid reversible formation of a complex between the oxidant and reductant, which later decomposes in a slow step by an inner-sphere electron transfer process, was observed. Acetaldehyde, benzaldehyde and CO2 were detected as the products. The oxidation path proceeds via a C-C bond cleavage. An increase in the acidity and a decrease in the solvent polarity increased the rates. The formation of free radicals was identified by a positive acryl amide polymerization test. The formation constants and unimolecular decomposition rate constants for the complexes were separated. From these kinetic observations the mechanism suggested was as given in scheme 25 taking mandelic acid as a typical example:
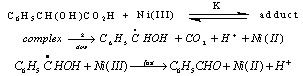 | Scheme 25. Reaction between mandelic acid and Ni(III) |
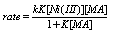 | (37) |
3.4.2.3. Reactivity of Ethanolamines with Nickel(III) in Aqueous Solution: Control by Enthalpy and Entropy of Activation [64].
- Ethanolamine, diethanolamine, and triethanolamine react with Ni(III) in aqueous solution via an inner-sphere electron transfer mechanism. The orders in [substrate] and [oxidant] were fractional and one, respectively, suggesting the formation of a complex (K-path) between the substrate and the oxidant, which decomposes (k-path) in a rate-determining step. Thermodynamic and activation parameters of K and k paths were determined and discussed. Solvent immobilization due to generation of H+ in the rate-determining step leads to the negative activation entropies. The rates increased with increasing [H+]. The mechanism suggested was as shown in scheme 26:
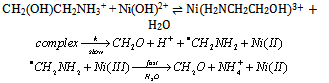 | Scheme 26. Reaction between ethanolamine and Ni(III) |
3.5. Outer-sphere Electron Transfer Reaction between Organic Oxidants and Inorganic Substrates: Oxidants are the Nitrobenzenes
- One electron reduction of nitrobenzenes by carbon dioxide radical anion (
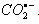 ) and hydrogen atom (H) [65]: During the course of characterization of nitrobenzene radical anions (RA-), nitrobenzenes were subject to the reaction of
) and hydrogen atom (H) [65]: During the course of characterization of nitrobenzene radical anions (RA-), nitrobenzenes were subject to the reaction of 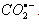 . The yields of RA- were characterized based on the thiocyanate (G(OH) + G(H) = 6.6) and methyl chloride (G(
. The yields of RA- were characterized based on the thiocyanate (G(OH) + G(H) = 6.6) and methyl chloride (G(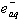 ) = 3.0)) dosimetries. Reactions of aqueous carbon dioxide radical anion (
) = 3.0)) dosimetries. Reactions of aqueous carbon dioxide radical anion (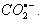 ) with p-substituted nitrobenzenes were studied by pulse radiolysis attached with time resolved uv absorption technique.
) with p-substituted nitrobenzenes were studied by pulse radiolysis attached with time resolved uv absorption technique. 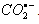 radicals were produced by the reaction of formate ion with primary radicals (OH) generated from water radiolysis in N2O saturated aqueous solutions. At pH 7 these radicals were quantitatively oxidized by nitrobenzenes to yield carbon dioxide and nitrobenzene radical anion (RA-) with second order rate constants (~ 2 X 109 M-1 s-1) close to little lower than the diffusion controlled limit (5 X 109 M-1 s-1) for almost all the twelve nitrobenzenes studied except the one with an electron donating substituent, the 4-methoxy nitrobenzene which reacted with a rate constant of 1.2 X108 M-1 s-1. The yield of RA- was found to be quantitative when studied spectroscopically compared to that obtained due to direct reaction of
radicals were produced by the reaction of formate ion with primary radicals (OH) generated from water radiolysis in N2O saturated aqueous solutions. At pH 7 these radicals were quantitatively oxidized by nitrobenzenes to yield carbon dioxide and nitrobenzene radical anion (RA-) with second order rate constants (~ 2 X 109 M-1 s-1) close to little lower than the diffusion controlled limit (5 X 109 M-1 s-1) for almost all the twelve nitrobenzenes studied except the one with an electron donating substituent, the 4-methoxy nitrobenzene which reacted with a rate constant of 1.2 X108 M-1 s-1. The yield of RA- was found to be quantitative when studied spectroscopically compared to that obtained due to direct reaction of 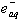 with nitrobenzene. And similarly H (produced from radiolysis of water) atoms were oxidized to hydrogen ions whose yields were found to be quantitative when compared to the CH3Cl dosimetry. The following is the reaction scheme 27.
with nitrobenzene. And similarly H (produced from radiolysis of water) atoms were oxidized to hydrogen ions whose yields were found to be quantitative when compared to the CH3Cl dosimetry. The following is the reaction scheme 27.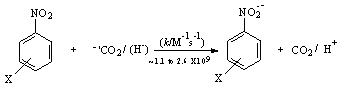 | Scheme 27. Reaction of carbon dioxide radical/hydrogen radical with nitro benzene |
3.6. Do Nitrobenzenes with Electron-Donating Substituents React with 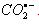 via Inner-Sphere Electron Transfer Reaction?
via Inner-Sphere Electron Transfer Reaction?
- One electron reduction of nitrobenzenes attached with electron-donating substituents by carbon dioxide radical anion (
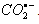 ) [65]: The investigations focused on this heading were not studied in detail in this direction in 1984 when this article was published in JACS [65] except for the determination of the optical properties like the extinction coefficients of nitrobenzene radical anions. While writing this review a gap was figured out for a reaction that involves an organic oxidant and inorganic substrate which could react via an inner-sphere electron transfer reaction. An insight in to the rate constant data revealed that the nitrobenzene with electron donating substituent, the 4-methoxy nitrobenzene reacted with
) [65]: The investigations focused on this heading were not studied in detail in this direction in 1984 when this article was published in JACS [65] except for the determination of the optical properties like the extinction coefficients of nitrobenzene radical anions. While writing this review a gap was figured out for a reaction that involves an organic oxidant and inorganic substrate which could react via an inner-sphere electron transfer reaction. An insight in to the rate constant data revealed that the nitrobenzene with electron donating substituent, the 4-methoxy nitrobenzene reacted with 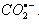 with far less than diffusion controlled limited rate constant. The rate constants for other nitrobenzenes like 4-hydroxy and 4-amino substituted nitrobenzenes could not be determined due to very low absorption of the radical anion and the overlap of the uv-vis spectrum of both the nitrobenzene and the radical anion. Had these rate constants been determined, some idea would have been evolved about the course of the reaction? But certainly these two nitrobenzenes should have reacted very slowly that can one take this scheme as an example of an inner-sphere electron transfer reaction between an organic oxidant and an inorganic substrate via an adduct? Fast response esr technique would have given an answer about the existence of the adduct if at all it had formed. To fill the gap and to under this heading in this review a hypothetical reaction has been suggested as shown in the following reaction scheme 28:
with far less than diffusion controlled limited rate constant. The rate constants for other nitrobenzenes like 4-hydroxy and 4-amino substituted nitrobenzenes could not be determined due to very low absorption of the radical anion and the overlap of the uv-vis spectrum of both the nitrobenzene and the radical anion. Had these rate constants been determined, some idea would have been evolved about the course of the reaction? But certainly these two nitrobenzenes should have reacted very slowly that can one take this scheme as an example of an inner-sphere electron transfer reaction between an organic oxidant and an inorganic substrate via an adduct? Fast response esr technique would have given an answer about the existence of the adduct if at all it had formed. To fill the gap and to under this heading in this review a hypothetical reaction has been suggested as shown in the following reaction scheme 28: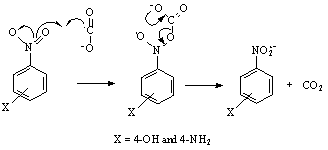 | Scheme 28. Reaction between carbon dioxide radical and nitro benzene via an adduct |
3.7. Inner-Sphere Electron Transfer Reaction between Organic Oxidants and Organic Substrates: Oxidants are the Nitrobenzenes.
3.7.1. One-Electron Reduction of Nitrobenzenes by α-Hydroxymethyl Radical via Addition/Elimination. An Example of an Organic Inner-Sphere Electron-Transfer Reaction [65]
- Reactions of aqueous α-hydroxymethyl radicals •CH2OH with p-substituted nitrobenzenes were investigated by using product analysis, in-situ-radiolysis-ESR spectroscopy, pulse radiolysis methods and also time resolved optical and conductance detection techniques. •CH2OH was produced by H abstraction from methanol by the OH (and H) radicals generated by irradiation of N2O-saturated aqueous solutions containing 0.1-2.0 M methanol. On addition of 0.1 to 1.0 mM para-substituted nitrobenzenes to the solutions, at pH 3-6 nitroxide type radicals were produced, as observed by in-situ-radiolysis-ESR experiments. These radicals were formed by addition of •CH2OH to the nitro group as shown in scheme 29. Nitrobenzene radical anions were not detected.
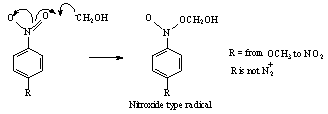 | Scheme 29. Reaction between α-hydroxymethyl radical and nitro benzene |
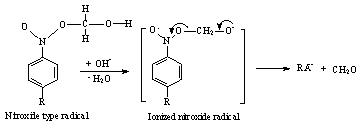 | Scheme 30. Reaction between α-hydroxymethyl radical and nitro benzene in presence of OH |
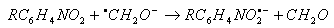 | (38) |
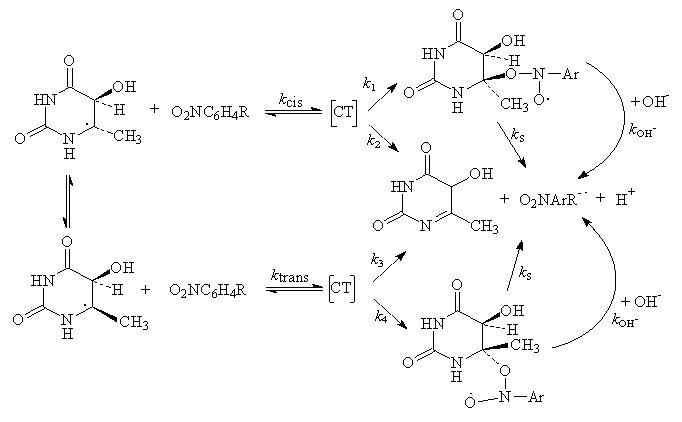
3.7.2. Reaction of 6-yl Radicals of Uracil, Thymine, and Cytosine and Their Nucleosides and Nucleotides with Nitrobenzenes via Addition to Give Nitroxide Radicals. Hydroxide ion-catalyzed Nitroxide Heterolysis [66]
- The 6-yl radicals produced by addition of OH to C(5) of the C(5)/C(6) double bond of naturally occurring pyrimidine bases, nucleosides, and nucleotides, or those formed by H-abstraction from C(6) of 5,6-dihydropyrimidines, react with para-substituted nitrobenzenes by addition to yield nitroxide-type radicals which were characterized by ESR and optical detection techniques. In basic solution the nitroxide radicals deprotonate: with the radicals derived from the free bases deprotonation occurs at N(1), with those from the nucleosides and nucleotides of uracil deprotonation at N(3) is observed. The ionized 6-yl radicals react with the nitrobenzenes also by addition with rate constants considerably higher than those for the case of the neutral radicals. The neutral nitroxide radicals react with OH- to give the nitroxide radical anion which was able to undergo a unimolecular heterolysis reaction and results in the formation of the nitrobenzene radical anion. In the case of the nitroxide radicals having N(1)-H (the radicals derived from the free bases), the OH- catalysis of nitroxide heterolysis proceeds via deprotonation at N(1) and the unimolecular rates are comparatively high. On substitution of N(1) by a Me or (deoxy)ribosyl(phosphate) group the site of OH- attack is changed to N(3)-H (with the uracils) or to N'(4)-H (with the cytosines). With radicals from N(1)-alkylated pyrimidines the rates of heterolysis of the nitrobenzene adducts are considerably lower than if N(1) carries a proton. The addition/OH- catalyzed elimination sequence of interaction of pyrimidin-6-yl radicals and nitrobenzenes results in the ultimate transfer of an electron from the pyrimidine radical to the nitrobenzene and was therefore an example of a one-electron redox reaction. The course of events was shown in scheme 31 where 4-NAP indicates 4-nitroacetophenone.
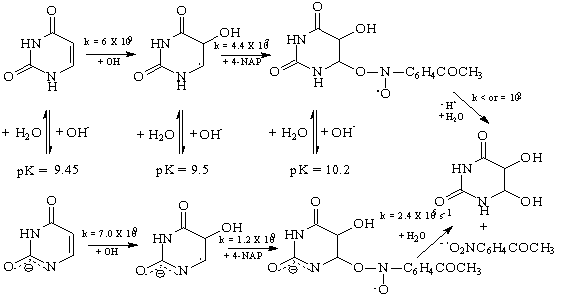 | Scheme 31. Reaction events of 4-NAP and pyrimidin-6-yl radical |
 | Scheme 32. Reaction events of α-hydroxy ethyl radical with nitro benzene |
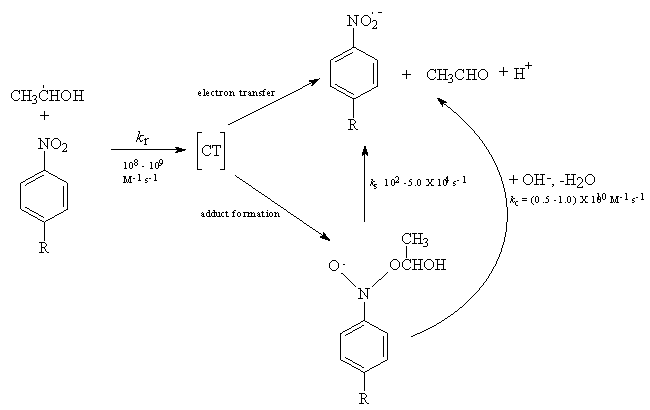
3.7.3. One-Electron Reduction of Nitrobenzenes by α-Hydroxy Ethyl Radical via Electron Transfer and Addition/ Elimination: Effect of Substituents on Rates and Activation Parameters for Unimolecular Heterolysis of Nitroxyl-Type Tetrahedral Intermediates [65]
- In the reaction of HO•CHCH3 with nitrobenzenes both addition and electron transfer took place, the fraction of electron transfer increasing with the electron-withdrawing power of the substituent in the nitrobenzene. Occurrence of both these reaction paths was successfully studied by time dependent absorption spectroscopy. The transient spectra clearly showed that the adducts absorbed at 280-300 nm and the radical anions at 330-350 nm. The nitroxyl-type adducts underwent a spontaneous uni-molecular heterolysis (ks) to give CH3CHO, H+ and nitrobenzene radical anion. The rate constants ks (from <102 to 5x104 s-1) for this heterolysis increased with the electron-withdrawing strength of the substituent if it was on the benzene, and decrease if the substituent was on the CH3 carbon of the nitroxyl. The heterolysis was characterized by low (5-10 kcal/mol) activation enthalpies and strongly negative (-5 to -25 e.u.) activation entropies, which originated from hydration of a proton in the transition state. From the effect on the activation parameters exerted by substituents on the electron-acceptor and -donor parts of the nitroxyl radical, the heterolysis proceeded via a push-pull mechanism, and was entropy controlled. Both Hammett and Taft equations obeyed for heterolysis reaction and yielded ρ (=1.4) when susbtituents were changed in nitrobenzene ring and ρ* (= -1.15) when substituents were changed in the alcohol moiety. From the isokinetic plot (β = 255 K), these reactions were supposed to be entropy controlled one. The reaction is shown in scheme 32.The formation of acetaldehyde was quantitative from GC analysis. In the ESR spectra the formation of nitroxyl type radicals could not be detected as in the case of •CH2OH. However by the use of time dependent optical absorption and conductivity detection techniques both the nitroxyl type radicals and radical anions were detected. With increase in pH the ks was found to increase according to the equation kobsd = ks + kc [OH-]. At pH > 10.5 the rates of formation radical anions became independent of pH but dependent on [nitrobenzene] and the simultaneous biphasic formation of nitroxyl type radicals and radical anions became monophasic with very high rates, which clearly showed that the ionized hydroxy ethyl radical i.e. CH3•CHO- (pK = 11.3) reacted by direct electron transfer.The reaction path of ks took place via a synchronous O-H and C-O bond breaking and via a solvent assisted deprotonation as shown in scheme 33:This was supported by studying the kinetic isotope effect on ks. The
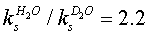 at 20oC using 4-nitrobenzonitrile with CH3•CHOH clearly showed that the above observation showing simultaneous breaking of N-O and C-O bonds in the transition state.
at 20oC using 4-nitrobenzonitrile with CH3•CHOH clearly showed that the above observation showing simultaneous breaking of N-O and C-O bonds in the transition state.3.7.4. One-Electron Reduction of Nitrobenzenes by •OH and H• Radical Adducts of 6-Methyluracil and 6-Methylisocytosine via Electron Transfer and Addition/Elimination: Effect of Substituents on Rates and Activation Parameters for Unimolecular Heterolysis of Nitroxyl-Type Tetrahedral Intermediates [67]
- The radicals formed by OH radical addition to C-5 of 6-methyluracil or of 6-methylisocytosine, i.e., 5-hydroxy- 5,6-dihydro-6-methyluracil-6-yl or 5-hydroxy-5,6-dihydro -6-methylisocytosin-6-yl, or that produced by H addition to C-5 of 6-methyluracil (or by H abstraction from C-6 of 5,6- dihydro- 6- methyluracil), i.e., 5,6 - dihydro-6 - methyluracil -6-yl, react in aqueous solution with para-substituted nitrobenzenes to give both nitrobenzene radical anions and nitroxyl-type radicals with rate constants that vary from 7 107 to (2-5) x 109 M-1 s-1, depending on the pyrimidine radical and on the nitrobenzene. The nitroxyl radicals undergo a spontaneous unimolecular heterolysis (ks) to yield (additional) nitrobenzene radical anion and oxidized pyrimidine with rate constants of 103 to 5 x 105 s-1, depending on the structure of the pyrimidine and of the nitrobenzene. This reaction is characterized by activation energies of 30-40 kJ mol-1 and by activation entropies of -7 to -89 J mol-1 K-1 (entropy control). The addition/elimination sequence constitutes a case of inner-sphere electron transfer reaction. The rate constants for the heterolysis reaction are a measure of the reducing power of 5,6-dihydro-6-methylpyrimidin-6-yl radicals. On this basis, the cytosine radicals are better reductants than the corresponding uracil radicals, and the radicals derived by H atom addition to pyrimidines are stronger reductants than those formed by OH radical addition. The experimental observations for studying these reactions were essentially similar to that studied with hydroxyl ethyl radicals and nitrobenzenes (Scheme 34).
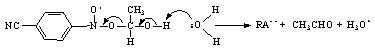 | Scheme 33. Spontaneous heterolysis of the α-hydroxy ethyl radical-nitro benzene adduct |
 | Scheme 34. Reaction of •OH and H• radical adducts of 6-methyluracil and 6-methylisocytosine with nitro benzene |
3.8. Outer-Sphere Electron Transfer Reaction between Organic Oxidants and Organic Substrate: Oxidants are the Nitrobenzenes
- In the oxidation of α-hydroxy isopropyl radical (CH3)2C•OH by nitrobenzenes [65], the nitroxyl radical of the type formed in the case of •CH2OH could not be detected and all nitrobenzenes oxidize (CH3)2C•OH by simple outer sphere electron transfer. The yield of acetone in this reaction was 86% and the remaining 14% was accounted for β-radicals, which has no reducing properties. Hence α-hydroxy isopropyl radicals were oxidized quantitatively. However a much weaker oxidant like 4-nitro-N,N-dimethyl aniline could not oxidize this radical by electron transfer, instead the course of the reaction was just similar to the events observed with that of •CH2OH. As the substituents in nitrobenzene become more and more electron withdrawing the reaction turned out to be addition/elimination and finally becomes pure electron transfer.
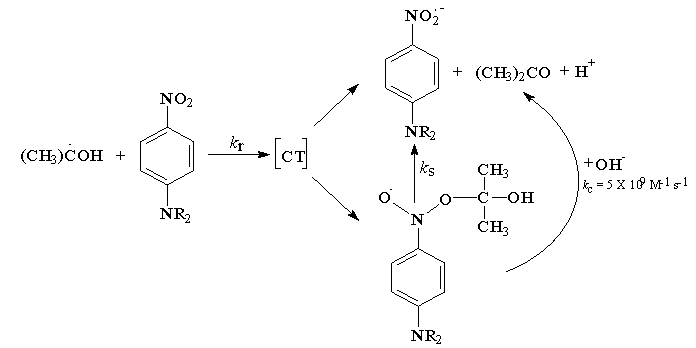 | Scheme 35. Reaction events of 4-nitro anile with (CH3)2C•OH |
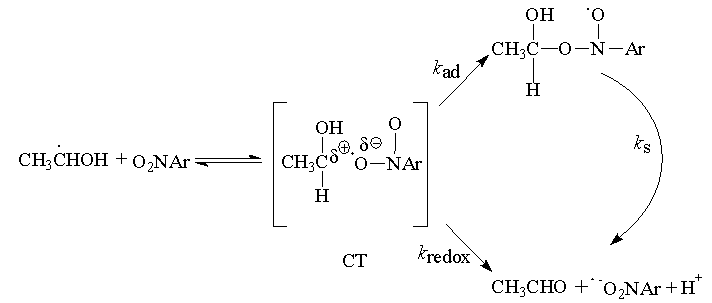 | Scheme 36. Reaction showing the CT-complex between ethanol radical and nitro benzene |
 ) between 4x107 and 2 x 109 M-1 s-1, i.e., below the diffusional level. The activation parameters for these reactions, which led to addition or to electron-transfer products, have been determined. The less than diffusion controlled rate constants were not due to large activation enthalpies but to very negative activation entropies [-10 to -80 J (mol K)-1]. In fact, the enthalpies (2 to 16 kJ mol-1) were in most cases below those for diffusion in water. The results were explained in terms of formation of a charge-transfer complex between the reactants and an ionic type transition state that results from electron transfer between the reactants as shown in scheme 36.Solvent immobilization caused to the ionic transition-state structures, leads to the negative activation entropies. The two different types of final products, i.e., nitroxyl and radical anion, are derived from the ion-pair-type transition state by collapse (giving addition/nitroxyl) or by separation by solvent (yielding electron-transfer/radical anion). In the hypothetical case as shown below, that the electron transfer was taking place followed by or concerted with proton transfer and (full) hydration of the proton, the electron would be trapped on the nitrobenzene for good, since hydration of the proton was irreversible due to enormous exothermicity of this reaction. In other words proton transfer would “freeze”, i.e. finalize electron transfer. (See scheme 37).
) between 4x107 and 2 x 109 M-1 s-1, i.e., below the diffusional level. The activation parameters for these reactions, which led to addition or to electron-transfer products, have been determined. The less than diffusion controlled rate constants were not due to large activation enthalpies but to very negative activation entropies [-10 to -80 J (mol K)-1]. In fact, the enthalpies (2 to 16 kJ mol-1) were in most cases below those for diffusion in water. The results were explained in terms of formation of a charge-transfer complex between the reactants and an ionic type transition state that results from electron transfer between the reactants as shown in scheme 36.Solvent immobilization caused to the ionic transition-state structures, leads to the negative activation entropies. The two different types of final products, i.e., nitroxyl and radical anion, are derived from the ion-pair-type transition state by collapse (giving addition/nitroxyl) or by separation by solvent (yielding electron-transfer/radical anion). In the hypothetical case as shown below, that the electron transfer was taking place followed by or concerted with proton transfer and (full) hydration of the proton, the electron would be trapped on the nitrobenzene for good, since hydration of the proton was irreversible due to enormous exothermicity of this reaction. In other words proton transfer would “freeze”, i.e. finalize electron transfer. (See scheme 37).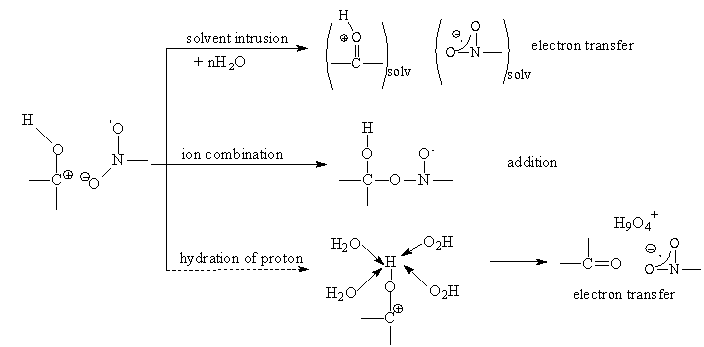 | Scheme 37 |
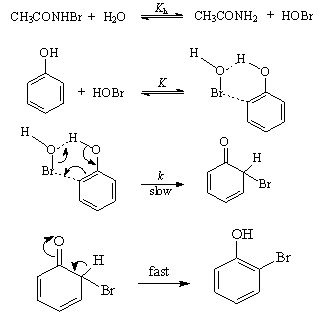
3.9. Oxidations by N-Bromoacetamide (NBA)
3.9.1. Kinetics and mechanism of oxidation of dimethyl sulfoxide by N-bromoacetamide (NBA): Outer-sphere oxygen atom transfer reaction [69]
- The title oxidation was carried out at constant [H+] and ionic strength. The order in [NBA] was unity. Pseudo first-order rate constants were calculated for various [DMSO]. The order in [DMSO] was found to be zero. An analysis of the rate dependence on [H+] at constant ionic strength revealed that the reactive species was found to be H2OBr+, formed in the rate-determining step by addition of H3O+ to HOBr. The rates were negligibly affected by added salts and dependent on solvent polarity. The sole product of the reaction was identified as dimethyl sulfone. The activation entropy for this reaction was compared with that of a NBA-alcohol reaction structurally related to DMSO. In the oxidation of iso-propanol (structurally related to DMSO) by NBA the activation entropy was found to be - 98.0 J (K mol)-1. This was explained by the production of H+ in the rate determining step by an α-H abstraction from the alcohol and this could be solvated extensively resulting in the freezing of free solvent molecules. But in the present reaction the small activation entropy of only -12 J (K mol)-1 was an indication that there was no such proton production in the rate determining step instead a proton was consumed liberating a free water molecule which could in fact lead to a positive entropy of activation. Based on the above arguments the mechanism suggested was as shown in scheme 38:
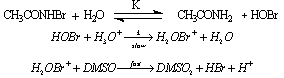 | Scheme 38. Reaction between DMSO and NBA |
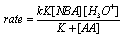 | (39) |
3.9.2. Kinetics and Mechanism of Oxidation of Benzaldehydes by N-Bromoacetamide in Acetic Acid-Water Mixture [70]
- The title oxidations in 40% acetic acid - water mixture were overall second-order, first-order in each [reactant]. The reactions were acid-catalyzed and the rate was not affected by the addition of Na2SO4. A plot of log k vs. Brown's
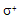 gave
gave 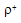 = -1.675 (r = 0.97) indicating an electron deficient center in transition state. However, p-nitro benzaldehyde deviated from the Hammett plot, its rate constant was found to be higher than expected. This might be due to the high degree of hydration of this compound compared to the other benzaldehydes. From the application of iso-kinetic relationship the reaction series was considered to be entropy controlled one. A probable mechanism involving formation of a cyclic intermediate in a fast step and subsequent decomposition in a slow step by a hydride transfer route was proposed as shown scheme 39:
= -1.675 (r = 0.97) indicating an electron deficient center in transition state. However, p-nitro benzaldehyde deviated from the Hammett plot, its rate constant was found to be higher than expected. This might be due to the high degree of hydration of this compound compared to the other benzaldehydes. From the application of iso-kinetic relationship the reaction series was considered to be entropy controlled one. A probable mechanism involving formation of a cyclic intermediate in a fast step and subsequent decomposition in a slow step by a hydride transfer route was proposed as shown scheme 39: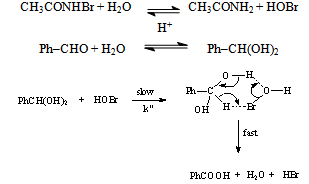 | Scheme 39. Reaction between benzaldehyde and NBA |
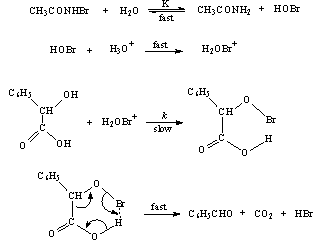
3.9.3. Kinetics and Mechanism of Uncatalyzed and Ruthenium(III) Catalyzed Oxidation of Mandelic Acid by N-Bromoacetamide in Aqueous Sulfuric Acid Medium [71]
- Rates of uncatalyzed and Ru(III)-catalyzed oxidation of mandelic acid (MA) by N-bromoacetamide (NBA) were measured in aqueous sulfuric acid. The overall order of the uncatalyzed reaction was one in each [reactant], i.e., NBA and MA. Under catalyzed conditions the order in [NBA] is unity, but that in [MA] and [Ru(III)] were fractional. The rates increase with increase in [H+] and decrease with increase in [acetamide]. The catalyzed redox process involves formation of an adduct between the catalyst and MA in a rapid reversible step, which later reacts with NBA biomolecularly in a slow step to give rise to products. Benzaldehyde was identified as the product from the 2,4-DNP test. Activation energy for catalyzed process (64.3 kJ mol-1) was less than the un-catalyzed reaction (77.3 kJ mol-1) indicating a good catalytic effect. The value of formation constant (K = 26.7 lit. mol-1) between Ru(III) and MA was much higher than the bimolecular rate constant (1.67 lit mol-1 min-1) in the un-catalyzed process, which indicated that Ru(III) interacts exclusively with MA in the formation of the adduct. This also got further support from the high negative entropy change (-59.0 J mol-1 K-1) for the formation of the adduct. From the above experimental facts the mechanisms for both the processes were as shown in scheme 40:
 | Scheme 40. Ru(III) catalyzed reaction between mandelic acid and NBA |
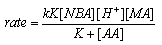 | (40) |
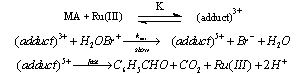 | Scheme 41. Reaction of mandeliacid with NBA in presence of Ru(III) |
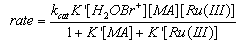 | (41) |
3.9.4. Kinetics and Mechanism of Oxidation of Aromatic Amines by N-Bromoacetamide [72]
- Kinetics of oxidation of anilines by N-bromoacetamide in methanol-water (50:50 vol./vol.) mixture had been investigated in the presence of mercuric acetate and excess acetamide. The total order of the reaction was two. Electron-releasing substituents in the aromatic ring accelerate the reaction rates and electron-withdrawing substituents retarded them. The value of the reaction constant
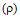 obtained from the Hammett plot (correlation coefficient. = 0.998) was -0.785. The product of the reaction was identified as the azobenzene. A mechanism shown in scheme 42 involving an electrophilic attack of the oxidant on the amino group had been proposed to explain the observed results.
obtained from the Hammett plot (correlation coefficient. = 0.998) was -0.785. The product of the reaction was identified as the azobenzene. A mechanism shown in scheme 42 involving an electrophilic attack of the oxidant on the amino group had been proposed to explain the observed results. 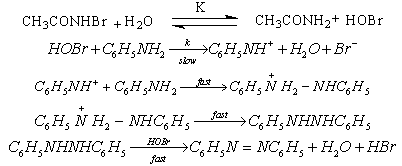 | Scheme 42. Reaction of anilines with NBA |
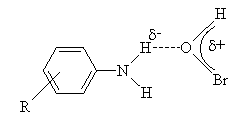 | Scheme 43 |
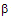 value of 0.186 indicating that there was extensive bond formation between aniline and HOBr in the transition state. The positive value of Brönsted coefficient and the negative value of Hammett’s reaction constant point out that the rate increased with the increase in basicity of the approaching nucleophile. From the iso-kinetic plot the reaction series was considered to be entropy controlled one.
value of 0.186 indicating that there was extensive bond formation between aniline and HOBr in the transition state. The positive value of Brönsted coefficient and the negative value of Hammett’s reaction constant point out that the rate increased with the increase in basicity of the approaching nucleophile. From the iso-kinetic plot the reaction series was considered to be entropy controlled one.3.9.5. Kinetics and Mechanism of Bromination of Phenols by N-Bromoacetamide: Effect of Substituents on Decomposition of Intermediate Adducts [73]
- The title reactions were studied in 40% aqueous methanol. A mechanism involving an intermediate six-membered cyclic transition state between phenol and HOBr (produced in situ by hydrolysis of N-bromoacetamide) was postulated for the title reaction as shown in scheme 44:
 | Scheme 44. Reaction of phenols with NBA |
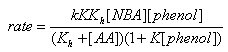 | (42) |
3.9.6. Kinetics and Mechanism of Bromination of Anisoles by N-Bromoacetamide in 50% Aqueous Acetonitrile [74]
- The rates of bromination of anisoles with HOBr produced in situ by the hydrolysis of N-bromoacetamide (NBA) have been measured in 50% aqueous acetonitrile in the presence of mercuric acetate and acetamide. The formations of an intermediate cyclic adduct between HOBr and anisole which decomposes in a slow step to yield the products has been proposed to explain the observed results. The thermodynamic parameters for the hydrolysis of NBA and adduct formation steps have been evaluated. The activation parameters for the first order decomposition of the adduct have also been calculated. The corresponding o-bromoanisoles have been identified as major components in the products. The kinetic trends, rate law and the mechanism were similar to those observed for phenols.
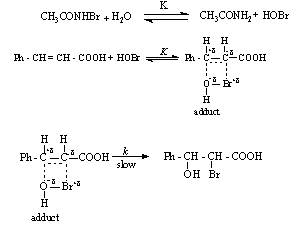
3.9.7. Kinetics and Mechanism of in situ Bromohydrination of Cinnamic Acids by N-Bromoacetamide [75]
- Bromohydroxylation of cinnamic acids (CA) by N-bromoacetamide (NBA) was studied in aqueous methanol containing Hg(OAc)2 and acetamide. The influence of substituents on the decomposition rate constant of the adduct was investigated. The corresponding bromohydrins were identified as the reaction products. The mechanism suggested was as given in scheme 45:
 | Scheme 45. Reaction of cinnamic acids with NBA |
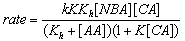 | (43) |
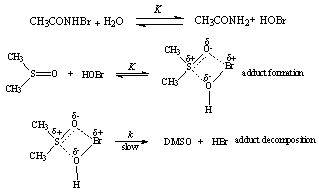
3.9.8. Solvent Effects on Kinetics of Oxidation of Dimethyl Sulfoxide by N-Bromoacetamide. An Example of Inner Sphere Oxygen Atom Transfer Reaction [76]
- Rates of oxidation of DMSO by N-bromoacetamide (NBA) in neutral aqueous solution were measured in the presence of acetamide. The order in [DMSO] was one below 0.01 mol dm-3, fractional at [DMSO] 0.01- 0.5 mol dm-3, and zero at [DMSO] > 0.5 mol dm-3. Different rate laws were operative under these three conditions, though HOBr was the effective oxidizing species in all the cases. The influence of the variation of solvent composition on the reaction rate was studied by using MeOH-H2O of various compositions. Thermodynamic parameters for NBA hydrolysis and adduct formation between HOBr and DMSO were evaluated. The activation parameters for the first-order decomposition of the adduct were also calculated for solvent compositions of 0-80% vol. aqueous MeOH. The free energies of activation increase with the MeOH content of the medium, whereas the enthalpies and entropies of activation show a more complex, but partially compensating behaviour. The mechanism suggested was given below in scheme 46 for the fractional order condition with DMSO:
 | Scheme 46. Reaction of DMSO with NBA |
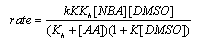 | (44) |
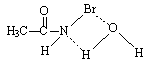 | Scheme 47. Interaction of water with NBA |
3.10. Oxidations by HOBr
- Survey of literature revealed that hypobromous acid (HOBr) is generated by hydrolysis of N-bromo compounds such as N-bromoacetamide (NBA), N-bromosuccinamide (NBS), and N-bromobenzamide (NBB), which later reacts with substrates. We have recently identified that HOBr was found to be easily generated in situ from NaBrO3 in the presence of an appropriate reducing agent such as NaHSO3, which was successfully used, in the synthesis of several organic compounds [77]. And we are the first to under take the kinetic investigations using this reagent. It was very easy to handle this reagent in aqueous solution as compared to that of the aqueous solutions of several N-halo compounds. Since the aqueous solutions of N-halo compounds are not stable, every time they have to be prepared afresh when ever necessary instead it was very easy, less time consuming and economical to handle the sodium bromate-sodium bisulfite reagent for generating HOBr. In view of the above advantages of sodium bromate-sodium bisulfite reagent over N-halo compounds, this reagent has been selected and used in the kinetic investigations of the reactions with DMSO [78], cinnamic acids [79], benzylamines [80], phenols [81] and benzaldehydes [82]. All these reactions followed the same kinetic trends mentioned as in the oxidation by NBA except that there was no reaction step involving the acetamide generation in the hydrolysis of NBA.
3.10.1. Kinetics and Mechanism of Oxidation of Dimethyl Sulfoxide by Sodium Bromate-Sodium Bisulfite Reagent in Aqueous Medium [78]
- Rates of oxidation of DMSO by HOBr produced in situ from sodium bromate - sodium bisulfite reagent were studied iodometrically in aqueous medium. The order in [DMSO] is one when [DMSO] < 0.01 mol dm-3, fractional when [DMSO] is 0.01-0.5 mol dm-3 and zero when (DMSO) > 0.5 mol dm-3. Different rate laws were operative under these three conditions though HOBr was the effective oxidizing species in all the cases. A mechanism involving an intermediate four-membered cyclic transition state between DMSO and HOBr with a formation constant (K), which decomposes in a slow step with a rate constant (k) was proposed. Thermodynamic parameters for the formation of adduct and activation parameters for the first-order decomposition of the adduct were evaluated. Activation parameters were also determined while the orders in [DMSO] were unity and zero. And all these were discussed. The reaction product was identified as di-methyl sulfone (DMSO2).
3.10.2. Kinetics and Mechanism of in situ Bromohydroxylation of Cinnamic Acids by Sodium Bromate-Sodium Bisulfite Reagent in Aqueous Acetonitrile Medium [79]
- Rates of bromohydroxylation of 4-substituted cinnamic acids with HOBr, produced in situ from sodium bromate-sodium bisulfite reagent have been studied in aqueous acetonitirile medium iodometrically. Thermodynamic parameters for the formation of the adduct and the activation parameters for the first order decomposition of the adduct have been evaluated in the temperature range and discussed. The influence of the substituents on the first order decomposition rate constant (k/s-1) of the adduct has been investigated. The corresponding bromodydrins of cinnamic acids have been identified as the reaction products.
3.10.3. Kinetics and Mechanism of Oxidation of Benzylamines by Sodium Bromate-Sodium Bisulfite Reagent in Aqueous Medium [80]
- Kinetics of oxidation of benzylamines (BA) by HOBr produced in situ from sodium bromate-sodium bisulfite reagent has been studied in aqueous medium at constant pH = 9.2 iodometrically. The reaction was overall second order and first order in each reactant i.e., HOBr and BA. HOBr has been postulated as effective oxidizing species. The activation parameters for the second order rate constant (k/dm3 mol-1 s-1) have been evaluated and discussed and they indicated the reaction series was entropy controlled. The influence of the substituents has been investigated. The Hammett ρ value was found to be – 0.64. A mechanism (scheme 48) involving transfer of a hydride ion from the methylene group of benzylamine to oxidant HOBr in the rate-determining step has been proposed. The sole reaction products have been identified as corresponding benzaldehydes and NH3.
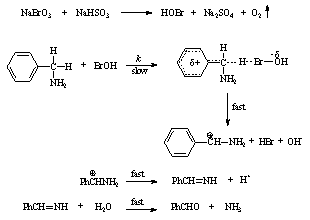 | Scheme 48. Reaction of benzylamines with HOBr |
3.10.4. Kinetics and Mechanism of Bromination of Phenols by Sodium Bromate-Sodium Bisulfite Reagent in Water -Acetonitrile Mixture [81]
- Kinetics of bromination of phenols with HOBr produced in situ from sodium bromate-sodium bisulfite reagent has been studied in water-acetonitrile mixture iodometrically. The order of the reaction was found to be unity in [HOBr] and fractional in [phenol]. HOBr has been established as the effective brominating species. A mechanism involving a six-membered cyclic adduct between phenol and HOBr, which decomposes in a slow step has been proposed. Thermodynamic parameters for the adduct formation and the activation parameters for the first order adduct decomposition have been evaluated and discussed. The influence of the substituents on the first order decomposition rate constant (k/s-1) of the adduct has been discussed. The sole reaction product has been identified as the corresponding o-bromophenol.
3.10.5. Kinetics and Mechanism of Oxidation of Benzaldehydes by Sodium Bromate-Sodium Bisulphite Reagent in Aqueous Acetonitrile Medium [82]
- Kinetics of oxidation of substituted benzaldehydes (BH) by HOBr produced in situ from sodium bromate-sodium bisulphite reagent has been studied iodometrically in aqueous acetonitrile (50:50 v/v) medium at constant acidity. The reaction was overall second order and first order in each reactant. The reaction was acid catalyzed and the rate was not affected by the addition of Na2SO4. Formation a cyclic intermediate in a slow step and subsequent decomposition of this in a fast step by a hydride ion transfer route were suggested as a probable mechanism. Activation parameters have been evaluated and discussed. The influence of the substituents on the oxidation reaction rates and rate constants has been investigated. The sole reaction product has been identified as the corresponding benzoic acid.
3.11. Oxidations by Peroxo Compounds: Two Peroxo Anions, Peroxodiphosphate [83] and Peroxomonosulfate [84]
3.11.1. Kinetics and Mechanism of Ruthenium(III) Catalyzed Oxidation of Lactic Acid by Peroxodiphosphate [83]
- The kinetics of Ru(III)-catalyzed oxidation of lactic acid (LA) by peroxodiphosphate (PP) was studied in aqueous acid medium. An analysis of the rate dependence on [H+] revealed that the active oxidizing species in the oxidation could be H3P2O8-. The Ru(III) catalysis of (PP-La) reaction was explained in terms of a 1:1 complex formation between Ru(III) and LA which later reacts with PP bimolecularly to give acetaldehyde and carbon dioxide as the products. The mechanism suggested was as given in scheme 49:From the above mechanism the rate law (eqn. 45) derived was:
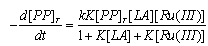 | (45) |
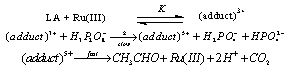 | Scheme 49. Reaction peroxodiphosphate with lactic acid |
3.11.2. Reactivities of Mono and Di-Anions of Peroxomonosulfuric acid Towards Benzaldehydes: A Kinetic and Mechanistic Study [84]
- The rates of reactions of different species of peroxomonosulfate (PMS) with benzaldehydes RC6H4CHO (R = H, 4-NO2, 3-NO2, 4-Me, and 4-MeO) have been measured iodometrically in CH3CN-water in the presence of phosphate buffers. The reaction was overall second order and the rate increased with the increase in pH and was very fast and almost instantaneous at pH > 8. This could be due to the slow accumulation of more reactive PMS2- (see below). Hence any reaction beyond this pH could not be conducted. Neutral peroxomonosulfuric acid reacts very slowly with benzaldehydes. The locus of the Hammett plot had a minimum near the point for benzaldehyde showing that there was a change in the rate-determining step. A mechanism involving addition/elimination between PMS- and benzaldehyde near neutral pH has been proposed as shown in Scheme 50:
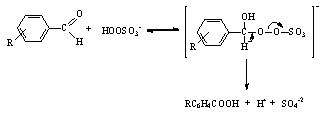 | Scheme 50. Reaction of benzaldehyde with peroxo mono sulfuric acid |
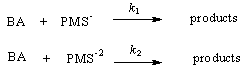 | Scheme 51. Reactions of anions of PMS with benzaldehyde |
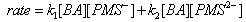 | (46) |
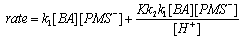 | (47) |
3.12. Inner-Sphere Synchronous Three Electron Transfer Reactions
- A review has been published on this aspect [85] along with some original publications of the work [86, 87], and [88]. In literature there were several examples on Cr(VI) oxidations. An interesting feature of this reaction was that Cr(VI) oxidation of a mixture of a hydroxy acid or di-carboxylic acid and 2-propanol proceeds much faster than that of either of the two substrates alone. A termolecular complex was supposed to decompose in a rate-determining step [86]. Free radicals were detected as intermediates by polymerization technique. Acetone from 2-propanol and CO2 from acid were the products. The individual oxidation proceeds by two- electron oxidation and the mixed system proceeds by three- electron oxidation and this is now known as “co-oxidation”. A study of mixed substrate oxidation of an alcohol and an aldehyde showed that the redox process took place in a single step three-electron oxidation [87]. Another important aspect of this kind of study was that a substrate having three functional groups of same kind or different could also proceed in a single step three-electron oxidation [88].
3.12.1. Co-Oxidation of Iso-Propanol and Lactic Acid in the Presence of Chromic Acid: A Case of Three-Electron Oxidation Study [86]
- The rate of oxidation of (CH3)2CHOH by chromic acid was increased considerably in the presence of lactic acid. The rates were acid catalyzed and dependent on the polarity of solvent but independent of added salts. A mechanism in which formation of a 1:1:1 termolecular complex followed by its decomposition in a single 3-electron redox reaction was suggested. However, the acceleration in rate was not observed when 1,2-glycols were used in place of LA indicating the absence of “co-oxidation”. This might be due to the fact that 1,2-glycols are not good complexing agents like the one, hydroxy acid or a di-carboxylic acid. From the products and the kinetic trends the mechanism was suggested as given in scheme 52:
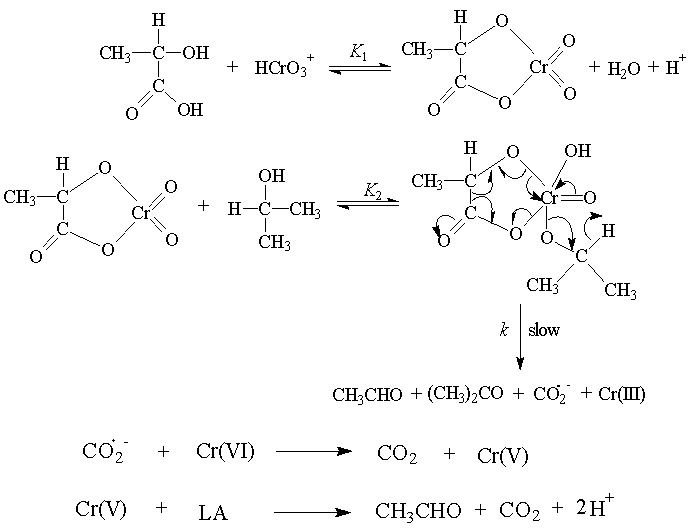 | Scheme 52. Reaction of 2-propanol with Cr(VI) in presence of lactic acid |
3.12.2. Three-Electron Oxidation: Chromic Acid Oxidation of a Mixed Substrate System of an Alcohol and Aldehyde [87]
- The kinetics of oxidation of CH3CH2CHO (I) and (CH3)2CHCH2OH (II) and their mixture by chromic acid was examined in aqueous acetic acid. A first-order dependence on [Cr(VI)] was observed. First-order kinetics in [I] or [II] alone were also found; fractional orders were found for I and II when their mixture was oxidized. The rates were independent of added salts and changes in solvent polarity. A mechanism was proposed involving neutral molecules and formation of a di-ester (shown in scheme 53) of chromic acid with aldehyde and alcohol in two successive equilibrium steps. The di-ester then decomposes in a slow step to give the products. Equilibrium constants and the rate constants for the uni-molecular decomposition of the di-ester were evaluated.
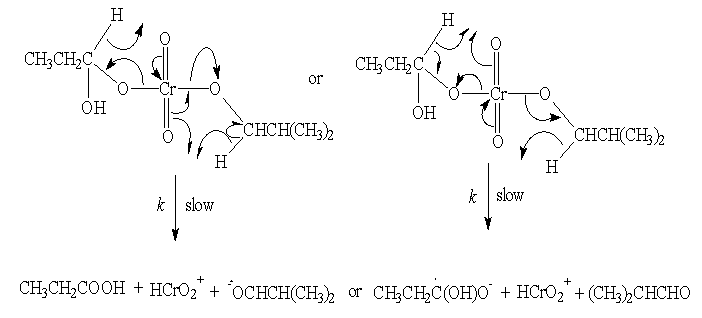 | Scheme 53. Reaction of mixture of an alcohol and an aldehyde with Cr(VI) |
3.12.3. Kinetics and Mechanism of Oxidation of Glycerol by Chromic Acid in Sulfuric Acid Medium [88]
- The title reaction was overall second order, first order in each [reactant]. The reaction was acid-catalyzed and the rates were independent of added salts, but dependent upon solvent polarity. The oxidation products were HCHO and CHOCHO, formed via a free radical mechanism in direct three electron change step. The oxidation of glycerol was compared with that of 1,2-diol and 1-propanol. From the change in activation entropy, it was found that the large negative value for glycerol oxidation (- 164 J K-1 mol-1; but for 1,2-diol and 1-propanol oxidation they were only -98.0 and - 90.0 respectively) could lead to suggest the formation of a rigid binary complex (Scheme 54) in the rate determining step which latter gives the products. Here glycerol was supposed to act as a tridentate ligand with its three OH groups.
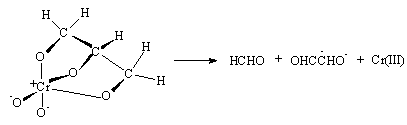 | Scheme 54. Decomposition of the binary complex of Cr(VI) |
3.13. Inner-Sphere Synchronous Four Electron Transfer Reaction: A Rapid Reaction of Manganese(VII) with Two Component Substrate Systems Containing 2-Propanol and some Bifunctional Compounds: A Kinetic Study[89]
- The rates of the permanganate ion oxidation of some bifunctional compounds such as hydroxy acids and 1,2-glycols in the presence of 2-propanol (IPA) were higher than the rates of oxidation of either of those compounds or IPA alone under identical conditions. The orders in [substrate] and [IPA] were found to be fractional. A probable mechanism involving a 1:1:1 termolecular complex (shown in scheme 55) and its decomposition in a single four electron transfer step was proposed as shown below taking lactic acid [LA] as a typical example to explain the kinetic results.
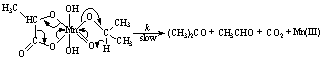 | Scheme 55. Decomposition of the complex of Mn(VII) |
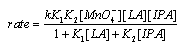 | (48) |
4. Conclusions
- Organic chemistry and inorganic chemistry investigated by the methods and techniques of Physics and Physical Chemistry have been particularly fruitful areas of study. And although physical chemists have always investigated the properties and reactions of organic compounds, the discipline that today we recognize as Physical Organic Chemistry really began when organic chemists themselves became seriously interested in the relationship between molecular structure and chemical properties, and in how, at the molecular level the reactions of organic compounds take place. This review in my opinion may lead the readers to offer to the students at an early stage the important distinction between experimental facts and reaction mechanisms.Structures of reactants and products are usually the principal features of a chemical reaction to be established. Equilibrium constants for reversible processes are also normally determinable, and a third experimental aspect of a reaction which may be quantified is its rate (or velocity). Molecular structures and reaction parameters can be expressed numerically with associated degrees of precision which are usually taken as measures of accuracy. In contrast a mechanism - a view of how reactants become products - constitutes a set of deductions derived by theory and intuition from the experimental results. A mechanism is not, therefore, a primary experimental feature in the way that a molecular structure, an equilibrium constant, or a rate constant is, and we can never express it in fine detail and with complete confidence.The general organic chemistry text book may not describe completely how reactions take place, why one reactant is more reactive than the other under the same experimental conditions. It is aimed in this review, however, is not to demonstrate in detail, but how experimental results could lead towards a mechanistic understanding of a particular reaction. This may not be possible over a wide subject area. But indeed it is the aim of the author to illustrate in a single progressive account the application of principles by which this knowledge is used to interpret the experimental results with in the general context of organic chemistry. This attempt could lead to the idea of interfacing between organic and physical chemistry may be seen as an amalgamation.Dedication: This review is dedicated to Prof. Dr. Steen Steenken (Retd) of the then Max-Planck-Institute for Radiation Chemistry, Muelheim, a.d Rhur, Germany.Acknowledgement: It is my pleasure to owe my heartfelt gratitude and sincere thanks to Dr. B. K. Mishra, M.Sc., Ph.D., D.Sc., Professor and Head, Department of Chemistry, Sambalpur University, Jyoti Vihar, Orissa-768 019, India and to Professor G. B. Behera, M.Sc., Ph.D., D.Sc., (Retd.), Department of Chemistry, Sambalpur University, without these two good personalities, my review would have not come into existence for which I was dreaming since a long time.I should not forget to thank Professor Dr. D. Schulte-Frohlinde, (Retd, now living in USA) the then Director, Max-Planck-Institute for Radiation Chemistry, Muelheim a.d. Ruhr, Germany for having invited me as a Post-Doctoral Fellow during 1980-88 and attaching me to a giant Physical Organic Chemist Professor Dr. Steen SteenkenI take this opportunity to convey my sincere gratitude to Professor Dr. Steen Steenken (Retd), Max-Planck-Institute for Bioinorganic Chemistry (The then Max-Planck-Institute for Radiation Chemistry), Muelheim a. d. Ruhr, Germany.I also thank Professor John P. Richard and Professor Tina L. Amyes (then at the Department of Chemistry, University of Kentucky, Lexington, Kentucky) Department of Chemistry, State University of New York at Buffalo, Buffalo, U.S.A. for their good wishes.I thank all my PhD students Dr. P. Musala Reddy, Dr. T. Rayapa Reddy, Dr. S. Venkateswarlu, Dr. S. Venkateswar Rao and Dr. T. Satyanarayana Reddy, Readers, Department of Chemistry, Sardar Patel college, Secunderabad from where I also started earning my bread and butter through teaching chemistry during 1978-1983, Dr. (Mrs.) P. R. Sharadamani, Reader, Department of Chemistry, Kasturba Gandhi College for women, Secunderabad, Dr. Yogyaraj, Lecturer, Department of Chemistry, N. B. Science College, Hyderabad, Dr. G. Krishna Reddy, Postgraduate Teacher in Chemistry, then at Kendriya Vidyalaya, Kanchanbagh, Hyderabad, Dr. R. Sanjeev, as a DST (New Delhi) funded JRF, Dr. J. Viroopakshappa, Reader, Department of Chemistry, Tara Government Degree College, Sangareddy (Andhra Pradesh) were instrumental in contributing a lot to my research program since last 37 years.Thanks are also due to Professor Masaaki Mishima and Professor Yuho Tsuno, Department of Chemistry, Faculty of Science, Kyushu University, Fukuoka, Japan, Dr. Yong Gu Lee, a Post-Doctoral Fellow from the Department of Chemistry, Kangwon National University, Chuncheon, Korea, and Dr. Douglas J. Rice, then a Graduate Student in Dr. John’s laboratory, in the Department of Chemistry, University of Kentucky, Lexington, Kentucky who contributed their share to my research program.I express my thanks to my wife Vijaya, my son Suresh, and my daughter Sharada for their patience and uncomplaining tolerance during my entire research activity.Finally I wish to record my indebtedness to my father Akkaiah (Late) and to my mother Savithramma who gave me the birth to exist in this world in my present position.I have written this review with utmost care not to involve any grammatical mistakes taking the help of my friends, who teach English, but some mistakes will inevitably remain; these are my own responsibility.