-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2012; 2(2): 18-22
doi: 10.5923/j.chemistry.20120202.04
An atomic-level Catalysis of Aun(SR)m Nanoclusters for Stereoselective Epoxidation of Stilbene
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLYan Zhu 1, Guoxiang Chen 1, Rongchao Jin 2, Yuhan Sun 1
1Low Carbon Conversion Center, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201203, P. R. China
2Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania, 15213, USA
Correspondence to: Yuhan Sun , Low Carbon Conversion Center, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201203, P. R. China.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The efficient and stereoselective epoxidation of cis-stilbene and trans-stilbene was achieved at the atomic level using atomically precise Aun(SR)m nanoclusters catalysts in mild conditions, involving thiolate-capped Au25(SR)18, Au38(SR)24, and Au144(SR)60 nanoclusters. These Aun nanoclusters, which are ideally composed of an exact number of gold atoms, have unique atom packing structures and non-metallic electronic properties and show super catalysis comparing to current gold nanoparticles. The atomic-level understanding of mechamism is particularly interesting and the correlation of their structure and properties may provide us a deep insight into the fundamental nanogold catalysis.
Keywords: Aun Nanoclusters, Stereoselective Epoxidation, Atomic-Level Catalysis
Article Outline
1. Introduction
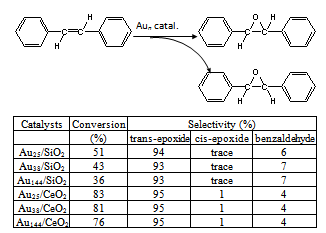
- The new development of structurally well-defined catalytic systems that emulate nature’s capacity to directly create stereochemical compounds is of great interest in the chemical synthesis era. Au nanoparticles have been proved to be a class of excellent catalysts for alkenes epoxidation implying propene, styrene, or cyclohexene epoxidation processes,1 however, the development of efficient stereoselective epoxidation still presents a great challenge, despite the steric demands of epoxides from non-terminal alkenes are readily differentiated for pharmaceuticals, agrochemicals, and fine chemicals. Hughes et al. reported the formation of trans- epoxide and cis-epoxide from the selective oxidation of cis-stilbene on Au/C catalyst in the presence of TBHP, whereas its application is only restricted in a small-scale laboratory experiment process by the very low yield (5.5% in toluene as a solvent).2 Additionally Lignier et al. found the high activity of Au/TiO2 in the stereoselective epoxidation of trans-stilbene in methylcyclohexane as a solvent, however the mechanistic description was concentrated on the effect of methylcyclehexane in the free-radical reaction, and made no mention of the catalysis of gold catalyst on trans-stilbene epoxidation.3 Recently we disclosed the effective epoxidition of styrene using atomically precise Aun(SR)m nanoclusters as catalysts and proposed a understanding of the nanocatalysis of Aun nanoclusters at atomic level, wherein each active site on Aun surface conducts selective access to epoxidation. It is realized that the site-specific catalysis of Aun nanoclusters can be employed to transfer a variety of non-terminal alkenes for stereoselective epoxidation. Herein, we report the atomic level catalysis of Aun nanoclusters for efficiently stereoselective epoxidation of cis-stilbene and trans-stilbene. Such Aun nanoclusters are composed of metal core with a precisely controlled, specific number of gold atoms, formulated as Aun(SR)m, where, n and m represent the respective numbers of gold atoms and thiolate ligands in the particle. Here, atomically monodisperse Aun(SR)m nanoclusters (where R stands for −CH2CH2Ph), involving Au25(SR)18 (~1 nm in diameter), Au38(SR)24 (1.3 nm in diameter), and Au144(SR)60 (1.6 nm in diameter) were synthesized by wet chemistry approaches.4-7
2. Experimental
2.1. Synthesis of Aun(SR)m catalysts (R= CH2CH2Ph)
- Au25(SR)18 nanoclusters: HAuCl4·3H2O (0.4 mmol, dissolved in 5 ml nanopure water), and tetraoctylammonium bromide (TOAB, 0.47 mmol, dissolved in 10 ml toluene), were combined in a 25 ml tri-neck round bottom flask. The solution was vigorously stirred for 15 min, and the aqueous was then removed. The toluene solution of Au (III) was purged with N2 and cooled down to 273 K in an ice bath over a period of 30 min under constant magnetic stirring. PhCH2CH2SH (0.17 ml) was added to the flask, and stirring was reduced to a very low speed (~50 rpm). After the solution turns to clear (~ 1 hr), NaBH4 (4 mmol, ~7 ml aqueous solution) was rapidly added all at once. The reaction was allowed to proceed overnight under N2 atmosphere. After aging overnight, ethanol was added to separate Au25 clusters from TOAB and other side-products. The Au25 clusters were collected after removing the supernatant. Au38(SR)24 nanoclusters: HAuCl4·3H2O (0.3 mmol) was first dissolved in methanol (20 mL) and phenylethanethiol (1.2 mmol, PhCH2CH2SH) was dissolved in nanopure water (10 mL). The two solutions were then mixed and then the mixture was cooled to ca. 273 K in an ice bath. NaBH4 (3 mmol, dissolved in 6 mL nanopure water) was added to the suspension under vigorous stirring. After reaction for another hour, the resulting precipitation (Au:SR) was collected by centrifugation and washed with methanol. The above Au:SR clusters were dissolved in H2O (4 mL). Then acetone (6 mL) and phenylethanethiol (8 mL) were added and the PhCH2CH2SH phase forms the upper layer and the aqueous solution of Au:SR clusters forms the bottom layer. The solution was heated to 353 K and allowed to react for several hours under vigorous stirring. After 3 h, the Au clusters were completely transferred to the organic phase. The isolated phenylethanethiol phase was washed thoroughly with excess ethanol and then with acetone to remove excess phenylethanethiol and byproducts. The major fraction containing Au38 clusters was extracted with toluene from the crude product. Au144(SR)60 nanoclusters: Toluene solution (20 mL) of TOAB (0.116 mmol) was mixed with an aqueous solution of HAuCl4·3H2O (0.1 mmol, 20 mL). After phase transfer of gold(III) into toluene, the aqueous layer was removed; PhCH2CH2SH (1.5 mmol) was added to the toluene solution, followed by stirring for 30 min. The mixture was then cooled to 0 oC. NaBH4 (0.1 mmol, dissolved in 20 mL 0 oC nanopure water) was rapidly injected into the solution under vigorous stirring. After reaction overnight, the organic phase was washed thoroughly with water and then rota-evaporated to dryness. The residue was washed with methanol to remove excess PhCH2CH2SH, TOAB, and other byproducts. The dried samples (~20 mg) were incubated in pure PhCH2CH2SH at 80℃ for 24 hr under vigorous stirring. The final solution was washed thoroughly with methanol and then acetone to remove by-products such as gold(I): PhCH2CH2SH complexes. The Au144(SCH2CH2Ph)60clusters were extracted from the products using a mixed toluene/acetone (1:1, v:v) solvent. Supported Aun catalysts: The supported Aun catalysts were obtained by stirring a solution of Aun(SR)m nanoclusters in dichloromethane with a calculated amount of SiO2 for one day and then the solution was removed in flowing nitrogen. The synthesis of Aun/CeO2 is similar with that of Aun/SiO2.
2.2. Catalytic Test
- Catalytic reaction was carried out at atmospheric pressure. Supported Aun catalysts (100 mg powder, 1%wt loading Aun(SR)m), stilbene (5 mmol), and TBHP (t-butyl hydroperoxide, 17 mmol) were added in a solvent (15 mL) in a 100 mL sealed glass reactor. Then the above mixture was heated to 82℃ under constant vigorous stirring in an oil bath for a certain reaction time. The solvent was removed with rotation evaporation and the products were analyzed by 1H NMR spectroscopy. The conversion and selectivity were calculated by quantitative NMR.
2.3. Characterization
- 1H NMR spectra were measured at Bruker AvanceTM 300 MHz. UV-vis absorption spectra (190-1100 nm) were recorded using a Hewlett-Pachard (HP) 8453 diode array spectrophotometer. Electrospray ionization mass spectra were acquired using a Waters Q-TOF mass spectrometer equipped with Z-spray source.
3. Results and Discussion
- These as-prepared Aun nanoclusters possess a non- crystallographic atom packing structures (Figure 1). In detail, the crystal structure of Au25(SR)18 nanocluster is arranged in six “V-shaped” –S(R)-Au-S(R)-Au-S(R)– staples, based on a centered icosahedral Au13 kernel encapsulated by twelve gold atoms shell.8,9 The entire particle adopts a quasi-D2h symmetry and is protected by 18 thiolate ligands. The crystal structure of Au38(SR)24 nanocluster is based on a face-fused biicosahedral Au23 core which adopts a D3h symmetry. The Au23 core is structurally strengthened by three monomeric staples –S(R)-Au-S(R)– distributed at the waist of Au23 and six dimeric staples –S(R)-Au-S(R)-Au-S(R)– evenly distributed on the two icosahedral units of Au23 core.10 The atom packing structure of Au144(SR)60 nanocluster is theoretically predicted to be arranged in –S(R)-Au-S(R)– staples with an icosahedral Au114 core capped by 30 gold atoms shell.11 Such precisely controlled Aun nanocatalysts offers unique opportunity for investigating nanocatalysis and the correlation of structure with their catalytic properties at the atomic level.
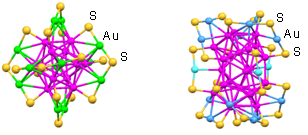 | Figure 1. The crystal structures of Au25 and Au38 nanoclusters. (for clarity, only Au and S atoms were shown) |
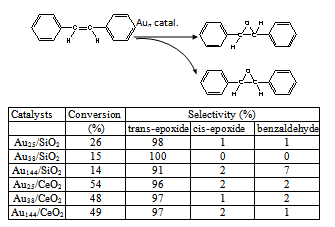
|
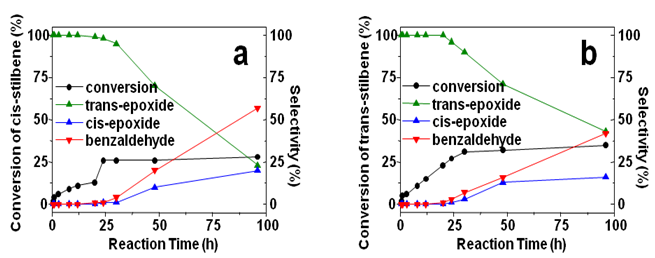 | Figure 2. The conversions and selectivities of (a) cis-stilbene and (b) trans-stilbene epoxidation on Au25/SiO2 catalyst at different reaction time, respectively |
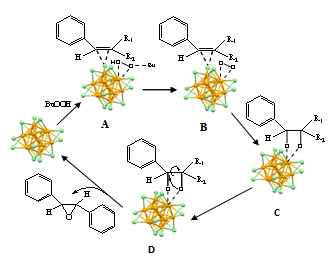 | Scheme 1. Proposed mechanism of the stereoselective epoxidation of stilbene on site-specific catalysis of Au25 nanocluster. Golden: Au atoms of the core; blue: Au atoms of the shell. (Where, when R1 is phene and R2 is H, the reagent is cis-stilbene; when R1 is H and R2 is phene, the reagent is trans-stilbene.) |
4. Conclusions
- In summary, we have developed a highly efficient stereoseletive epoxidation of stilbene using atomically precise Aun nanoclusters catalysts at an atomic level. The site-specific activity of Aun nanoclusters is attributed to their core-shell-type geometrical and electronic structures. The site-specific catalysis of Aun nanoclusters is promising to develop reaction classes that are widely viewed as important within the field of stereoselective catalysis.
Acknowledgements
- This work is financially supported by AFSOR, NIOSH and NSF.
References
| [1] | (a) T. Hayashi, K. Tanaka, M. Haruta, J. Catal., 1998, 178, 566; (b) A. Corma, H. Garcia, Chem. Rev., 2002, 102, 3837; (c) G. Grigoropoulou, J. A. Elings, Green Chem., 2003, 5, 1; (d) L. Cumaranatunge, W. N. Delgass, J. Catal., 2005, 232, 38; (e) A.S. K. Hashmi, G. J. Hutchings, Angew. Chem. Int. Et., 2006, 45, 7896; (f) P. Lignier, S. Mangematin, F. Morfin, J. L. Rousset, V. Caps, Catal. Today, 2008, 138, 50; (g) Y. Liu, H. Tsunoyama, T. Akita, T. Tsukuda, Chem. Commun., 2010, 46, 550. |
| [2] | M. D. Hughes, Y. Xu, P. McMorn, P. Landon, D. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin, C. J. Kiely, Nature, 2005, 437, 1132. |
| [3] | P. Lignier, F. Morfin, S. Mangematin, L. Massin, J. Rpusset, V. Caps, Chem. Commun., 2007, 186. |
| [4] | M. Z. Zhu, E. Lanni, N. Garg, M. E. Bier, R. C. Jin, J. Am. Chem. Soc., 2008, 130, 1138. |
| [5] | Z. K. Wu, C. Gayathri, R. R. Gil, R. C. Jin, J. Am. Chem. Soc., 2009, 131, 7220. |
| [6] | H. F. Qian, Y. Zhu, R. C. Jin, ACS Nano, 2009, 3, 3795. |
| [7] | H. F. Qian, R. C. Jin, Nano Lett., 2009, 9, 4083. |
| [8] | M. Z. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz, R. C. Jin, J. Am. Chem. Soc., 2008, 130, 5883. |
| [9] | M. Z. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. F. Qian, G. C. Schatz, R. C. Jin, J. Am. Chem. Soc., 2009, 131, 2490. |
| [10] | H. F. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer, R. C. Jin, J. Am. Chem. Soc., 2010, 132, 8280. |
| [11] | O. Lopez-Acevedo, J. Akola, R. L. Whtten, H. Gronbeck, H. Hakkinen, J. Phys. Chem. C, 2009, 113, 5035. |
| [12] | A. Corma, P. Atienzar, H. Garcia, J. Y. Chane-Ching, Nat. Mater, 2004, 3, 394. |
| [13] | A. Abad, A. Corma, H. Garcia, Top. Catal., 2007, 44, 237. |
| [14] | G. A. Somorjai, J. Y. Park, Angew. Chem. Int. Ed., 2008, 47, 9212. |