-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2012; 2(1): 28-32
doi: 10.5923/j.chemistry.20120201.07
Determination of Iron by Dispersive Liquid-Liquid Microextraction Procedure in Environmental Samples
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLF. Sánchez Rojas , C. Bosch Ojeda , J. M. Cano Pavón
Department of Analytical Chemistry, Faculty of Sciences, University of Málaga, Málaga, 29071, Spain
Correspondence to: F. Sánchez Rojas , Department of Analytical Chemistry, Faculty of Sciences, University of Málaga, Málaga, 29071, Spain.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
A greener analytical procedure based on dispersive liquid-liquid microextraction (DLLME) coupled to flame atomic absorption spectrometry (FAAS) detection was developed. Various influencing factors on the separation and preconcentration of iron, such as the acidity of the aqueous solution, extraction and disperser solvent type and their volume have been investigated systematically, and the optimized operation conditions were established. Under the optimum conditions i.e., pH 4.0, [DPTH] = 5x10-3%, a preconcentration factor of 45 was reached. The lower limit of detection (LOD) obtained under the optimal conditions was 9 μg L-1. The precision for 8 replicate determinations at 50 and 100 μg L-1 of Fe were 1.8 % and 4.4 % relative standard deviation (R.S.D.), respectively. The calibration graph using the preconcentration method was linear from 10 to 5000 μg L-1, with a correlation coefficient of 0.9904. The proposed method was successfully applied to the preconcentration and determination of iron in food, vegetation, soil and water samples.
Keywords: Iron, Flame atomic absorption spectrometry, Dispersive liquid–liquid microextraction, Water samples, Food, soil and vegetation samples
Cite this paper: F. Sánchez Rojas , C. Bosch Ojeda , J. M. Cano Pavón , "Determination of Iron by Dispersive Liquid-Liquid Microextraction Procedure in Environmental Samples", American Journal of Chemistry, Vol. 2 No. 1, 2012, pp. 28-32. doi: 10.5923/j.chemistry.20120201.07.
Article Outline
1. Introduction
- The accurate determination of trace elements in environmental samples is an important and challenging task in analytical chemistry. Direct determination of trace elements appears to be difficult task as the concentration of them is close to or below the detection limits of most of the analytical techniques besides the real sample matrix may cause serious interference during their determination process. However, these problems can be solved by applying the preconcentration techniques which can simultaneously remove the sample matrix and increase the concentration of analytes [1]. In this way, the direct determination of trace elements by spectroscopic methods, such as FAAS and ICP-OES, is often difficult because of insufficient sensitivity and selectivity of the used methods. For this reason, the preliminary separation and preconcentration of trace elements from the different matrices are required. Current trends in analytical chemistry are highly focused on improvement in the quality of analytical results, introduction of new technological developments with analytical use and, especially, miniaturization, simplification and automation of the whole analytical procedure. Recent research activities are being focused on the development of efficient, economical, and miniaturized sample preparation methods. Consequently different microextraction systems have been developed as solid-phase microextraction [2-4], cloud point extraction[5-7], dispersive liquid-liquid microextraction [8-10], etc. On the other hand, Fe is widely distributed in nature and is one of the most important elements in environmental and biological systems. Iron is the fourth most abundant element in the earth’s crust; it is present in a variety of rock and soil minerals. Concerning its biological activity, iron is a highly versatile element, serving as active center of proteins responsible for oxygen and electrons transference in metalloenzymes such as oxidases and dehydratases[11,12]. Therefore, it is very important to develop sensitive methods for quantitative determination of trace Fe in various matrices.In this work, a DLLME methodology has been developed and optimized for the extraction and determination of Fe. The method is based on chemical complexation of Fe(II) by 1,5-bis (di-2-pyridilmethylen thiocarbohidrazide) (DPTH). DLLME technique was used to extract the complex and FAAS was used to analyze the extracted product. The Fe was selected for evaluation of the procedure due to its environmental and biological importance. The FAAS method was used due to ease and low cost of operation. Using the developed method Fe can be analyzed in simple, rapid and inexpensive manner.Potential parameters affecting the DLLME and analytical performance are studied and optimized in detail, such as the type and volume of extraction and dispersive solvents as well as the extraction time. The proposed DLLME method has lower extraction time, since the extraction equilibrium is attained very quickly. DLLME has a wider linear range and lower solvent consumption. In addition to these advantages, DLLME is rapid, easy to use, inexpensive, and environmentally friendly. The proposed method was successfully applied to the preconcentration and determination of iron in environmental and food samples.
2. Experimental
2.1. Instrumentation
- Phase separation was achieved with a centrifuge Selecta Centromix in 10 mL calibrated conical tubes.A Varian Model SpectrAA 50 () flame atomic absorption spectrometer was used for the analysis with the appropriate iron hollow cathode lamp. The operating parameters were set as recommended by the manufacturer. Atomic absorption measurements were carried out in an air-acetylene flame. The following conditions were used: absorption line Fe: 248.3 nm; slit widths: 0.2 nm; and lamp currents: 4 mA.
2.2. Reagents and Samples
- High purity water (resistivity 18.2MΩcm) obtained by a Milli-Q® water purification system (Millipore, ) was used throughout this work. 1000 mg L-1 stock solutions of iron (E. Merck, ). Working standard solution was obtained daily by stepwise dilution of the standard stock solution. DPTH solution in N,N,-Dimethylformamide was prepared by dissolving solid reagent samples prepared and purified by the authors [13].The proposed method was evaluated by analysis of iron in several food, soil and plant samples. For this purpose, 0.9–1.8 g of diverse food, plant and soil were mineralized by microwave digestion, adjusted pH and diluted at convenient volume.Natural waters were collected in polypropylene bottles previously cleaned by soaking for 24 h in 10% (v/v) nitric acid and finally rinsed thoroughly with ultra-pure water before use.
2.3. Dispersive Liquid-Liquid Microextraction Procedure
- For DLLME under optimum conditions, 10 mL analyte solution containing iron, 2 mL acetate buffer solution pH 4, 0.5 mL of 0.1% DPTH solution in DMF as chelating agent and 0.2 mL of ascorbic acid was placed in a 15 mL screw cap glass test tube. Then, 1 mL of ethanol (as disperser solvent) and 200 µL of chloroform (as extraction solvent) were rapidly injected into a sample solution by using a microsyringe. A cloudy solution was formed in the test tube and separation of the phases was achieved by centrifugation at 3800 rpm for 5 min. After this process, a small droplet of organic phase was sedimented in the bottom of conical test tube. After removal of the whole aqueous solution, 50 µL of the extraction phase was diluted with 0.5 mL of 0.1M HNO3/ethanol to avoid very high signals obtained when measuring the absorbance of the organic phase directly, and the iron concentration was determined by FAAS by using a hollow cathode lamp of iron at 248,3 nm.
3. Results and Discussion
3.1. Optimization of the experimental conditions of the method
- Some preliminary experiments were carried out in order to investigate the quantitative extraction of Fe(II) ions using the DPTH reagent in the absence of other metal ions. The optimum conditions for the extraction of Fe(II) were established by varying the experimental parameters, such as pH of aqueous phase, ascorbic acid concentration, the concentration of ligand, solvent (disperser and extraction) type and volume, and ionic strength.
3.2. Effect of pH
- The pH is a very important parameter for the extraction studies. Much of the selectivity achieved in these extractions depends on adequate control of pH. The pH of the aqueous solution is an important factor in DLLME of Fe(II) using DPTH, because this parameter is directly related to the formation of metal–ligand species. In this work, the effect of the acidity of the aqueous solutions on the extraction of Fe(II) were examined in the pH range of 2.0–9.0 by using acetate or phosphate buffer and the results are shown in Figure 1. Thus, the value of pH 4.0 was selected for the following experiments.Also, the influence of acetate buffer solution amount was investigated for variation of volume added from 1 to 3 mL. The extraction efficiency was stable in all studied range. A volume of 2 mL of acetate buffer solution 0.2M was selected as optimum value for subsequent work.
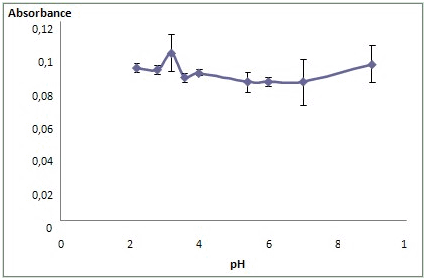 | Figure 1. Influence of pH |
3.3. Effect of ascorbic acid concentration
- By introducing ascorbic acid into the sample, the Fe(III) in the sample was reduced to Fe(II). Therefore, total Fe(II+III) in the sample was presented as Fe(II), and reacted with DPTH to form the Fe(II)-DPTH complex. The effect of the ascorbic acid concentration was investigated in the range of 0-0.04%. The optimum value selected was 0.02%.
3.4. Effect of chelating reagent (DPTH) concentration
- The effect of the ligand concentration was studied by extracting iron(II) ions with different amounts of the DPTH reagent. As shown in Figure final concentration of 5x10-3% was sufficient for optimum results. This concentration ensures sufficient excess to compensate for any consumption of the reagent by other metals.
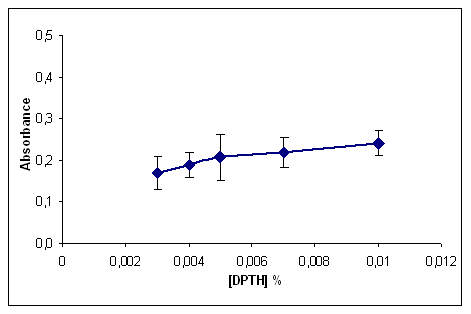 | Figure 2. Influence of reagent concentration |
3.5. Effect of ionic strength
- For investigating the influence of ionic strength on performance of DLLME, various experiments were performed by adding different amount of NaCl (0–1.5% (w/v)). Other experimental conditions were kept constant. No significant impact on the analytical signal was observed in the studied range enabling the possibility of utilizing the proposed method for saline samples.
3.6. Effect of type and volume of the disperser solvent
- The main criterion for disperser solvent in DLLME is its miscibility with both water and the extraction solvent. In this study acetone, methanol, ethanol, 1-propanol and 2-butanol were evaluated as disperser solvents. With 200 µL of chloroform and 1.5 mL of each disperser solvent, the analytical signals for iron with ethanol were comparable with that obtained with methanol and acetone and were higher than that obtained with1-propanol and 2-butanol (Figure 3). So, because of its lower toxicity, ethanol was selected as the disperser solvent.
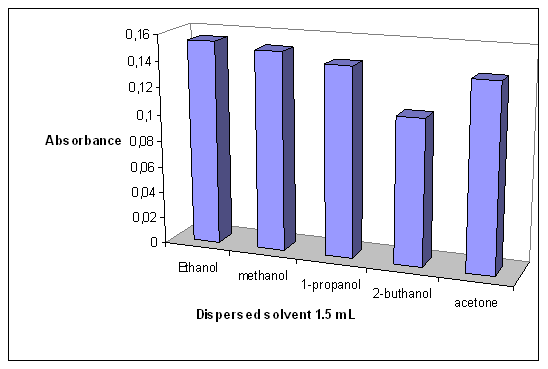 | Figure 3. Effect of type of the disperser solvent |
3.7. Effect of type and volume of the extraction solvent
- One of the most important factors affecting the extraction efficiency and enrichment factor of the metal complexes in DLLME is the nature of organic solvent and its volume. It should be higher density than water and have extraction capability of the interested compounds and low solubility in water. Several extracting solvents including chloroform, dichloromethane and carbon tetrachloride were investigated. The experiments were performed by using 200 µL of each extracting solvent and 1 mL of ethanol (as the disperser solvent). Results showed that the maximum extraction recovery was obtained by using chloroform.In order to examine the effect of the extraction solvent volume, solutions containing different volume of chloroform (150, 200, 250 and 300 µL) were subjected to the same DLLME procedure. When the volume of extraction solvent was increased, the volume available for the measurement also increased, but the enrichment factors decreased. Thereby, in the following studies, the optimum volume of 200 µL was selected for the extraction solvent volume.
3.8. Analytical performance
- Under the above optimum conditions, linear range, calibration graph, detection and determination limits, preconcentration factor and precision were obtained (Table 1). The calibration graph was linear in the range of 10–5000 µg L−1 of iron.
|
3.9. Application to real samples
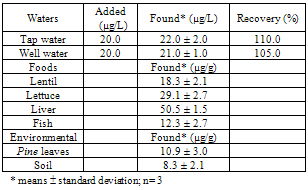
- The proposed DLLME–FAAS methodology was applied to the determination of iron in several water, food, soil and vegetation samples.In view of the application of the method to determination of iron in waters (tap water and well water) standard solutions containing iron were added to samples. Standard addition methods were used in all instances and the results were obtained by extrapolation. These results, as the average of three separate determinations, are shown in Table 3. The proposed method gave satisfactory average recoveries.The method was extended for the separation and determination of Fe(II) in real samples. The presented method was applied to the various food, vegetation and soil samples for the separation and enrichment of iron. The results were given in Table 3.
|
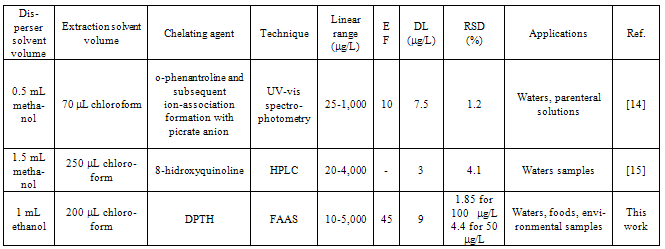
3.10. Comparison with other DLLME procedures for iron
- As far we know only two papers are published by DLLME procedure to determine iron based on the formation of the complexes. Comparisons are enclosed in table 4. From this table the proposed method presents detection limit in the order of previously papers and higher linear dynamic range.
|
4. Conclusions
- Sample preparation by DLLME is a procedure that considered inside the Green Chemistry, because of the small volumes of dissolvent employed.In this work, a simple, rapid, and sensitive DLLME concentration technique coupled with FAAS has been developed for the determination of iron in food, vegetation, soil and water samples.All variables that influence in the formation of the complex Fe–DPTH and then application of DLLME procedure have been optimized. Employing FAAS as detection technique, the detection limit obtained is in the order of µg L-1 that by the direct method of FAAS is impossible to obtain because of the low sensitivity that presents.To study the applicability of the proposed method, and have not samples certified, we have analyzed waters previously fortified, to check the percent of recovery, using the procedure of DLLME and by standard additions method.Food, vegetation and soil samples analyses have been made applying the proposed procedure and calculating the concentration of iron directly of the calibration curve.The results obtained demonstrated the applicability of the proposed method for this type of samples.
ACKNOWLEDGEMENTS
- The authors thank to the Ministerio de Ciencia e Innovación for supporting this study (Projects CTQ2009- 07858) and also the Junta de Andalucia.
References
| [1] | J. Minczenski, J. Chwastowska, R. Dybezynski, Separation and Preconcentration Methods in Inorganic Analysis, Ellis Horwood, Chichester, 1982 |
| [2] | A.R. Turker, Clean 35 (2007) 548 |
| [3] | K. Pyrzynska, Trends Anal. Chem. 29 (2010) 718 |
| [4] | S. Matsuokaa, K. Yoshimurab, Anal. Chim. Acta 664 (2010) 1 |
| [5] | C. Bosch Ojeda, F. Sánchez Rojas, Anal. Bioanal. Chem., 394 (2009) 759 |
| [6] | C. Bosch Ojeda, F. Sánchez Rojas, Microchim. Acta, 2012, DOI 10.1007/s00604-011-0717-x |
| [7] | C. Bosch Ojeda, F. Sánchez Rojas, J.M. Cano Pavón, Res. J. Pharm. Biol. Chem. Sci. 1 (2010) 514 |
| [8] | C. Bosch Ojeda, F. Sánchez Rojas, Chromatographia 69 (2009) 1149 |
| [9] | C. Bosch Ojeda, F. Sánchez Rojas, Chromatographia 74 (2011) 651 |
| [10] | F. Sánchez Rojas, C. Bosch Ojeda, J.M. Cano Pavón, Anal. Methods 3 (2011) 1652 |
| [11] | H. Bagheri, A. Gholami, A. Najafi, Anal. Chim. Acta 424 (2000) 233 |
| [12] | C.J. Chacarolli, J.F. Andrade, O.M. Guimaraes, V.R. Balbo, C.S. Venezuela, F.S. Teruel, Anal. Chim. Acta 411 (2000) 217 |
| [13] | J. Bonilla Abascal, A. Garcia de Torres, J.M. Cano Pavon, Microchem. J. 28 (1983) 132 |
| [14] | A.B. Tabrizi, J. Hazard. Mater. 183 (2010) 688 |
| [15] | M.A. Farajzadeh, M. Bahram, M.R. Vardast, J. Sep. Sci. 32 (2009) 4200 |