-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Research in Cell Biology
p-ISSN: 2326-1366 e-ISSN: 2326-1382
2014; 2(1): 1-7
doi:10.5923/j.cellbiology.20140201.01
Nitric Oxide Activates Plasma Membrane Insertion of the Betaine/GABA Transporter in Renal Epithelial Cells
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLStephen A. Kempson1, Bailey M. Anderson1, Moshe Levi2, Judith Blaine2
1Department of Cellular and Integrative Physiology, Indiana University School of Medicine, Indianapolis
2Department of Medicine, University of Colorado Denver, Aurora
Correspondence to: Stephen A. Kempson, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, Indianapolis.
| Email: |  |
Copyright © 2014 Scientific & Academic Publishing. All Rights Reserved.
In the renal medulla nitric oxide (NO) plays an important role in sodium and water homeostasis, and NO synthase isoforms are expressed in medullary nephron segments. These segments also produce NO in response to hypertonic NaCl, but a possible role for NO during osmotic stress is unclear. When Madin-Darby canine kidney (MDCK) epithelial cells were switched from isotonic to hypertonic medium containing 1 mM nitroprusside, the transport activity of the renal betaine/GABA transporter (BGT1) was increased by 96% within 4 hr compared to hypertonic controls. BGT1 stimulation by nitroprusside also was observed after prior hypertonic adaptation for 24 hr. NO donors also stimulated the hypertonic activation of the system A amino acid transporter, SNAT2. Nitroprusside activation of BGT1 was not reproduced by membrane–permeable analogs of cyclic GMP. To understand the mechanism MDCK cells were transfected with EGFP-tagged BGT1 and imaged by total internal reflection fluorescence microscopy. The rate of recovery of plasma membrane fluorescence after photobleaching was increased almost two-fold in the presence of 1 mM nitroprusside, compared to controls. This suggests NO may accelerate the insertion of BGT1 protein into the basolateral plasma membrane during hypertonic stress, which helps to fully activate the adaptive response.
Keywords: Kidney, MDCK cells, BGT1, TIRF microscopy, NO, Confocal, Osmolyte
Cite this paper: Stephen A. Kempson, Bailey M. Anderson, Moshe Levi, Judith Blaine, Nitric Oxide Activates Plasma Membrane Insertion of the Betaine/GABA Transporter in Renal Epithelial Cells, Research in Cell Biology, Vol. 2 No. 1, 2014, pp. 1-7. doi: 10.5923/j.cellbiology.20140201.01.
1. Introduction
- Hypertonic stress in the kidney induces numerous insults and cellular adaptation is a complex process that goes beyond volume regulation. For example, high extracellular NaCl induces oxidative stress in kidney cells in culture [1, 2], and in medullary thick ascending limbs in situ [3], as well as DNA strand breaks in cultured cells and in renal medulla in vivo [4]. Cellular responses include cell cycle arrest, accumulation of heat shock proteins, and activation of mechanisms that detect, repair, and protect against hypertonicity-induced DNA damage [5]. Recent work in C. elegans worms indicate that additional responses may be involved [6]. It has been suggested that hypertonic stress may generate a combination of signals that together lead to full activation of the adaptive responses [5].One well-studied adaptation to chronic hypertonic stress in the kidney inner medulla involves cellular accumulation of organic osmolytes that do not disturb cell function. Betaine is present in plasma at 30-50 M in humans [7], and is derived mainly from the diet but can be augmented by synthesis in the liver and kidney [8]. It is freely filtered and reabsorbed by Na+ and H+ cotransport systems in the luminal membrane of the proximal tubule [9]. Betaine, together with myo-inositol, sorbitol, and glycerophosphorylcholine, serves as an organic osmolyte that is accumulated by cells of the inner medulla of animal and human kidneys to maintain osmotic balance [7, 10]. Intracellular betaine levels were determined to be at least 60 mM in tissue from inner medulla and papilla [10, 11]. Accumulation of betaine by medullary cells is mediated by the Na+ and Cl- dependent betaine/GABA transporter (BGT1), a member of solute carrier family 6 (SLC6A12) that also accepts GABA as a substrate [12]. Both synthesis and trafficking of BGT1 protein are upregulated by hypertonic stress and the protein is inserted in the basolateral plasma membrane of renal medullary cells [12]. Basolateral sorting is directed primarily by signals embedded within the primary structure of proteins [13], and both C-terminal trafficking motifs [14] and phosphorylation at T40 [15) have been shown to be critical for accurate BGT1 trafficking. Up- and down-regulation of BGT1 by changes in extracellular osmolarity is slow, requiring almost 24 hr. However, we and others have previously demonstrated relatively rapid regulation (within 30 min) of BGT1 transport in renal MDCK cells in response to extracellular ATP, adenosine, calcium and changes in extracellular pH [12]. NO production in the renal medulla plays a very important role in sodium and water homeostasis and the long-term control of arterial pressure [16, 17]. NO is highly diffusible and short-lived, and is synthesized from the amino acid L-arginine in a reaction catalyzed by NO synthase. Isoforms of NO synthase are found throughout the nephron [18] and NO production was detected directly in isolated thick ascending limbs using the fluorescent dye 4-amino-5-methylamino-2’,7’-difluorofluorescein diacetate (DAF-FM) [19]. NO action can be mediated by intracellular pathways that are cyclic-GMP dependent and by pathways that do not require cyclic GMP [18, 20]. Using microelectrodes, we showed previously that NO production by mouse medullary slices was initially 0.1 μM and increased in response to hypertonic bathing solution [21]. However the specific role of NO during osmotic stress is unclear. The present study is focused on the acute effects of NO donors, namely sodium nitroprusside (NP) and diethylamine NONOate (NONO), on the adaptive response of BGT1 to hypertonic stress in cultured renal medullary MDCK epithelial cells. Two different NO donors were tested to ensure that any response was due to released NO and not a side effect of the specific donor. The donors were tested both during the early phase (0 – 4 hr) and after completion (24 hr) of adaptation to hypertonic stress. The MDCK cell line was chosen because it produces NO [22, 23] and expresses BGT1 [15].
2. Materials and Methods
- Sodium nitroprusside (NP) and diethylamine NONOate sodium salt (NONO), from Sigma-Aldrich (St Louis, MO) were freshly prepared for each experiment as 100 mM solutions in phosphate buffered saline (pH 7.4). Stocks were used immediately by adding aliquots directly to experimental solutions (pH 7.4). Nitro–L-arginine methyl ester (L-NAME) from Santa Cruz Biotechnology was used as a non-specific inhibitor of NO synthase isoforms.MDCK cells (#CCL-34, American Type Culture Collection, Rockville, MD) were grown in monolayers in DMEM/Hams F12 (1:1) growth medium containing 10% bovine calf serum, penicillin (100 IU/ml) and streptomycin (100 μg/ml), and were maintained in an atmosphere of 5% CO2 in air. Cells were used between passages 15 and 40 and were grown on glass-bottom 35 mm culture dishes (Matek, Ashland, MA) for microscopy and in plastic 24-well plates for transport studies. Normal isotonic growth medium was made hypertonic (550 mOsm) by addition of NaCl crystals, and the osmolarity was determined by freezing point depression using an osmometer, as used and described previously [24].MDCK cells were pre-loaded with the fluorescent dye DAF-FM diacetate (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions. Briefly, MDCK cell monolayers on glass (Matek) were incubated with 10 uM DAF-FM diacetate for 60 min at 37℃ in isotonic DMEM growth medium containing 0.5% BSA but lacking both serum and phenol red to avoid interference with the fluorescent signal. The cells were washed to remove excess probe. The DAF-FM released by esterase activity remains intracellular and reacts specifically with NO to form a fluorescent product that is insensitive to pH changes [25, 26]. The live cells were covered with fresh DMEM medium, with or without addition of 1.0 mM NP, and were imaged immediately by confocal microscopy using 495 and 515 nm for excitation and emission, respectively, as described previously [24]. Quantitation of intracellular calcium was obtained using the calcium-sensitive fluorescent indicator Fura 2 exactly as described in our previous studies [27].The transport activity of BGT1 was measured as Na+-Cl--dependent uptake of [3H]GABA, or [14C] betaine by MDCK cells in monolayer culture, as in previous studies [27, 28]. Both substrates were tested to ensure uniformity of responses. The Na+-dependent system A amino acid transporter (SNAT2, SLC38A2) [29] was used as a control because, like BGT1, it is localized to the basal-lateral plasma membrane of MDCK cells and can be removed by endocytosis. Unlike BGT1, it is upregulated much more rapidly (within 5-6 hr) by hypertonic stress [28]. Transport activity of SNAT2 was determined as Na+-dependent uptake of [14C] methylaminoisobutyric acid, a derivative of alanine [28]. The basolateral surface of MDCK cells was imaged by total internal reflection fluorescence (TIRF) microscopy [30]. The system, used in our previous studies [31, 32], was an Axiovert 200 inverted microscope (Zeiss) equipped with a TIRF laser illumination system (Zeiss), a high numerical aperture (1.45) oil immersion 100x objective, and a Photometrics QuantEM camera, all under the control of Axiovision software (Zeiss). The microscope was equipped with an incubation chamber (37℃) with humidity and CO2 controlled atmosphere. The system was adjusted for TIRF imaging of the basal plasma membrane with 488 nm laser excitation according to the manufacturer’s instructions. The MDCK cells were grown on glass-bottom 35 mm culture dishes and the cells expressed BGT1 tagged with enhanced green fluorescent protein (EGFP), as described in our previous experiments [24]. The EGFP-tagged protein (EGFP-BGT1) was shown previously to behave and function identically to native BGT1 In MDCK cells [24]. Prior to use some cells were adapted to hypertonic growth medium (550 mOsm) overnight while controls remained in normal isotonic medium. Cells were switched to fresh hypertonic or isotonic medium, respectively, 4-5 h prior to imaging. All fresh media contained 0.1% bovine serum albumin but lacked serum and phenol red. When used, NP was added to hypertonic growth medium 15 min prior to imaging. A bleach step with 100% laser power for 5 min eliminated any fluorescence already present in the plasma membrane. The rate of fluorescence recovery after photobleaching (FRAP) was determined by collecting approximately 25-30 images at 75 sec intervals, using the 488nm laser at 5% power for 250 msec. Axiovision software was used to quantitate the fluorescence of individual cells in the collected images and rates of recovery were determined by linear regression analysis.Data are mean + SD of 3-4 experiments. Where appropriate, significant differences between groups (p<0.05) were determined by Student’s t-test or by analysis of variance and Tukey’s test for multiple comparisons. All images are representative of several cells on each coverglass.
3. Results
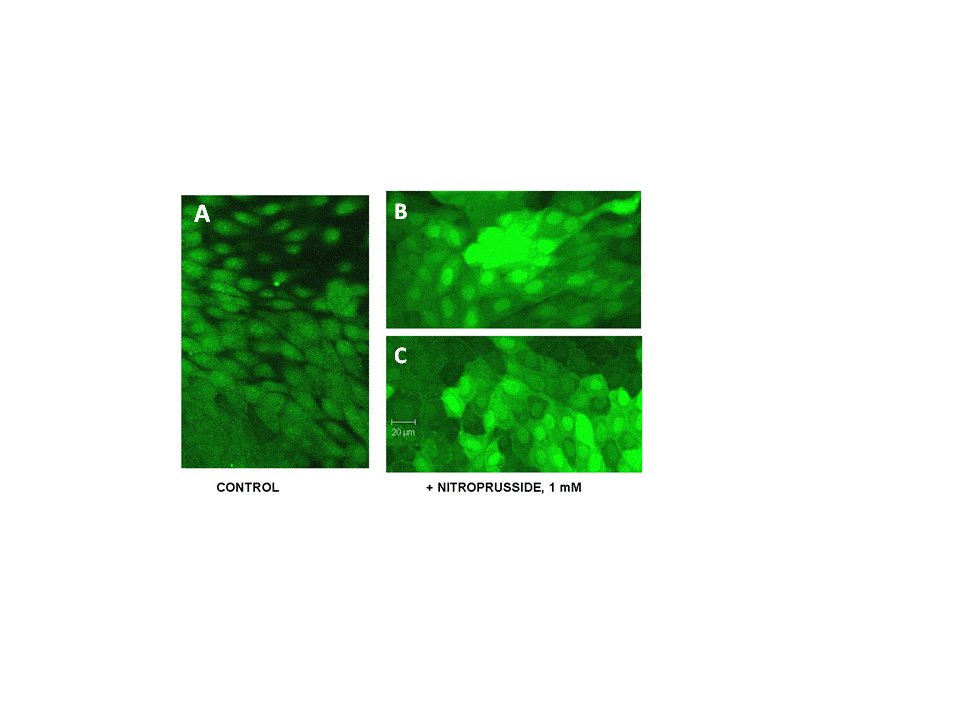 | Figure 1. As expected, addition of sodium nitroprusside (1.0 mM final concentration) to native MDCK cells for 1 h produced a marked increase in intracellular NO content. This was confirmed in live cells pre-loaded with the fluorescent dye DAF-FM diacetate. There was a marked increase in intracellular fluorescence after addition of nitroprusside (B,C) compared to controls (A) |
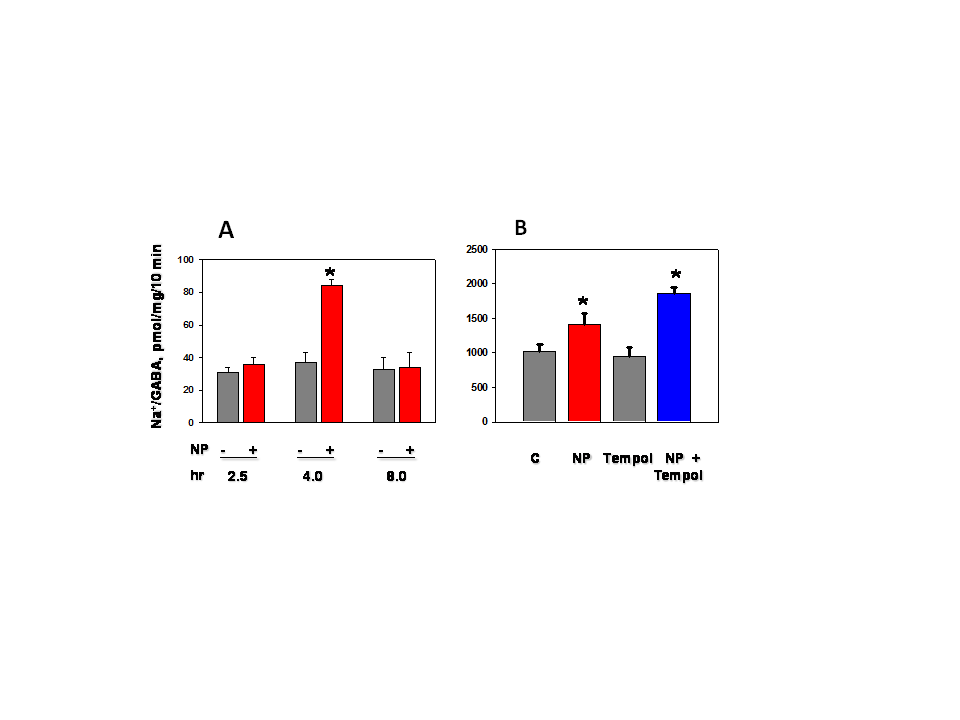 | Figure 2. A. Sodium nitroprusside (NP, 1.0 mM final concentration) activated BGT1 transport of GABA in MDCK cells within 4 hr when added at the onset of hypertonic stress (550mOsm). B. Similarly, NP also activated BGT1 transport when added for 2 h following 24 h of hypertonic stress, and this effect was enhanced by the presence of Tempol (1.0 mM). Controls (C) were exposed only to hypertonic stress. *Significant difference (p<0.05) compared to respective controls (n=3) |
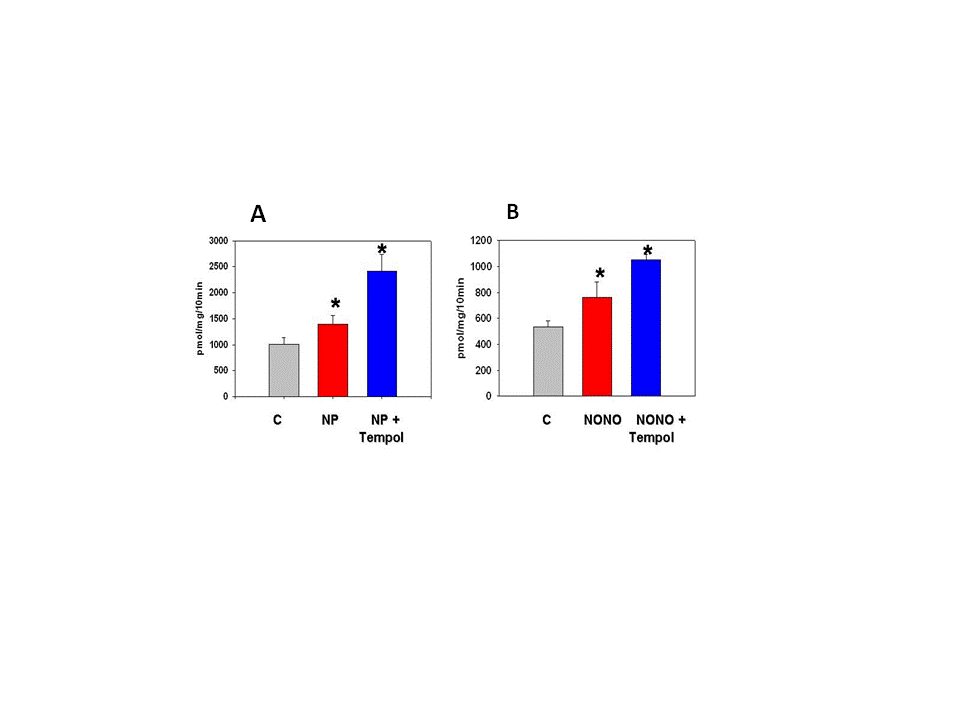 | Figure 3. A. NP (1.0 mM) also activated cell uptake of methylaminoisobutyric acid (substrate for SNAT2) after just 2 hr hypertonic stress. This effect was increased in presence of 1.0 mM Tempol. B. NONO (0.1 mM) reproduced the effects of NP. *Significant difference (p<0.05) compared to controls (n=3) |
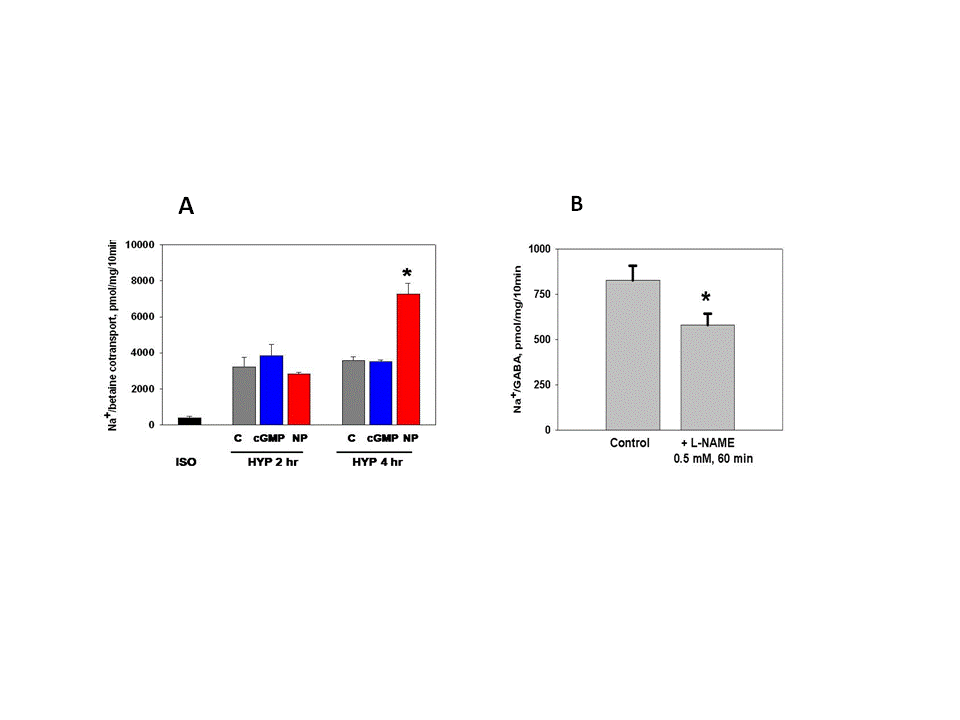 | Figure 4. A. 8-Bromo-cyclic GMP (cGMP, 0.2 mM final concentration) did not activate BGT1 transport of betaine during hypertonic stress (HYP) for 2 or 4 h, suggesting that activation by NO donors did not utilize cyclic GMP-dependent pathways. ISO, isotonic controls. B. Following upregulation by 24 h of hypertonic stress, BGT1 transport of GABA was inhibited after 60 min treatment with L-NAME at 0.5 mM final concentration. *Significant difference (p<0.05) compared to respective controls (n=3-4) |
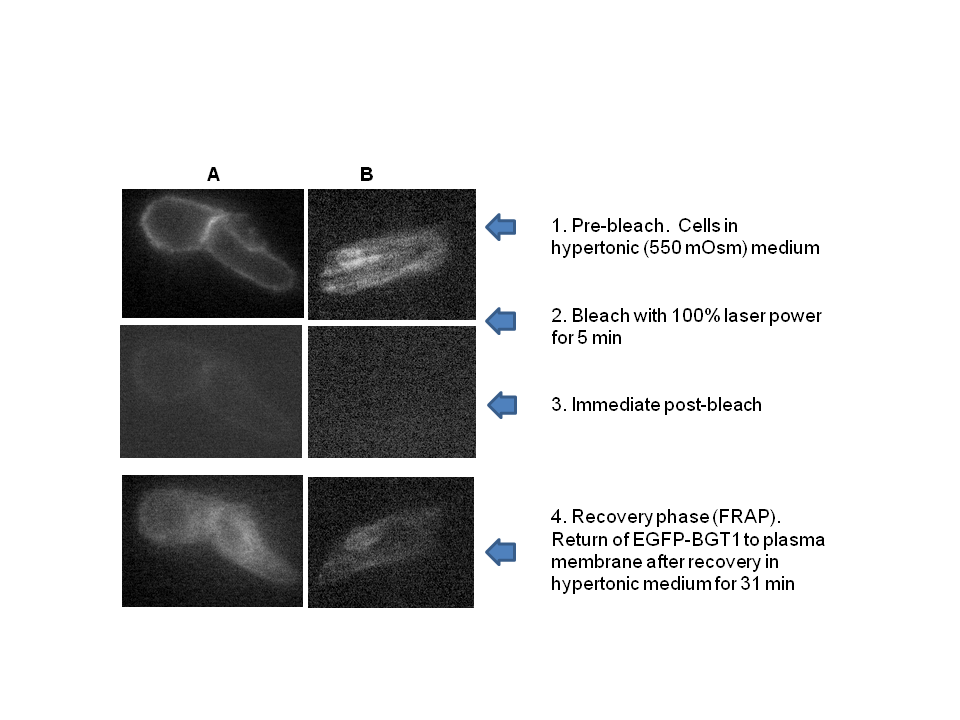 | Figure 5. Summary of the TIRF-FRAP technique for studying the basolateral plasma membrane of live MDCK cells. Two separate cells (columns A and B) are shown. (1) Following hypertonic adaptation, BGT1 tagged with EGFP was present in the plasma membrane. (2) Fluorescence was bleached with 100% laser power for 5 min. (3) Immediately after bleaching no membrane fluorescence was detected. (4) New EGFP-BGT1 was inserted in the plasma membrane in the fluorescence recovery phase (FRAP) during which 25-30 images were collected at intervals of 75 sec. Plasma membrane fluorescence showed significant recovery 31 min after photobleaching. Full details in Materials and Methods |
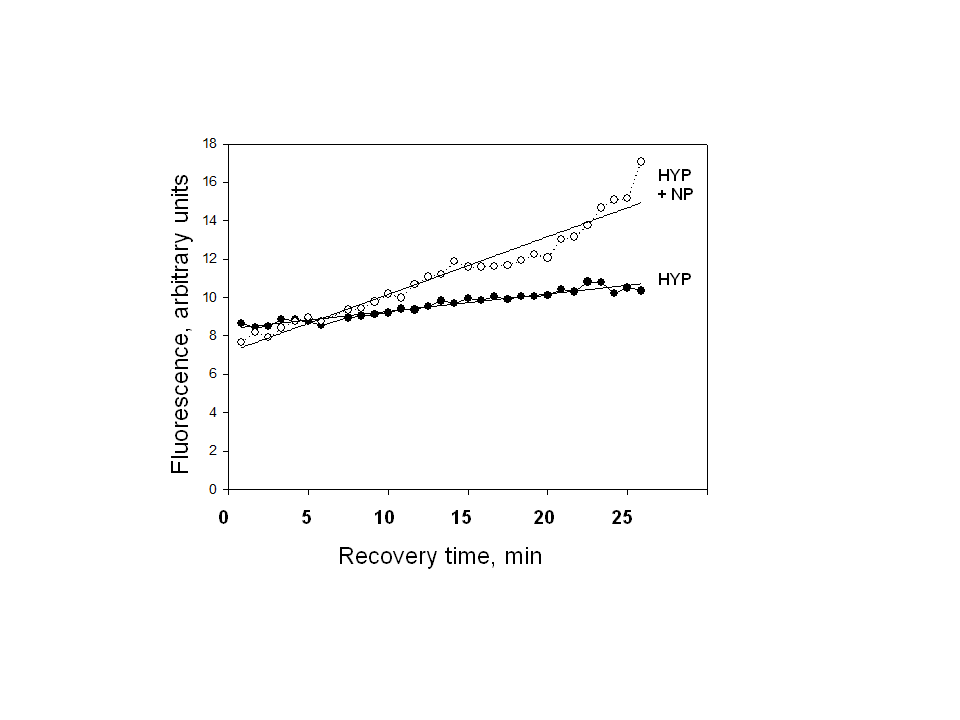 | Figure 6. Chronic hypertonic stress (24 h). Rate of EGFP-BGT1 fluorescence recovery in hypertonically (HYP) adapted MDCK cells was more than 3-fold in the presence of sodium nitroprusside (NP, 1.0 mM) compared to HYP controls. NP was added for 15 min prior to TIRF-FRAP imaging. Rates of recovery (fluorescence units/min), determined by linear regression, were 0.09 U/min (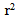 = 0.93) for HYP and 0.30 U/min ( = 0.93) for HYP and 0.30 U/min (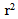 = 0.94) for HYP + NP = 0.94) for HYP + NP |
 | Figure 7. Acute hypertonic stress (4h). Rate of fluorescence recovery of EGFP-BGT1 after acute hypertonic stress was increased 3-fold compared to isotonic MDCK controls (ISO). Addition of NP (1.0 mM) produced an almost 2-fold additional increase in rate of recovery compared to acute hypertonic stress (HYP) alone. NP was added for 15 min prior to TIRF-FRAP imaging. Rates of recovery were 0.09 U/min ( = 0.98) for ISO, 0.27 U/min ( = 0.98) for ISO, 0.27 U/min ( = 0.96) for HYP, and 0.48 U/min ( = 0.96) for HYP, and 0.48 U/min (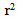 = 0.99) for HYP + NP = 0.99) for HYP + NP |
4. Conclusions
- We showed previously that NO production in medullary slices was increased rapidly (within 5 min) following onset of hypertonic stress [21]. This precedes the hypertonic activation of BGT1 transport which requires a few hours [24, 28]. NO likely accelerates the adaptive response to hypertonic stress in MDCK cells, specifically by upregulation of the BGT1 and SNAT2 transporters present in the basolateral plasma membrane. The intracellular signaling pathways that upregulate BGT1 remain to be elucidated but are not dependent on cyclic GMP since NP action was not reproduced by membrane-permeable analogs of cyclic GMP. Other intracellular pathways, possibly S-nitrosylation [23], may be involved. The final step in upregulation of BGT1 transport activity by NO likely involves increased insertion of BGT1 proteins into the basolateral plasma membrane. These data appear to conflict with a previous report that NO donors decreased expression of osmoprotective genes in MDCK cells by directly inhibiting the specific transcription factor termed tonicity-responsive enhancer binding protein (TonEBP) and also known as NFAT5 [12, 23]. However, the NO donors were applied for 24 h, a much longer period than used in the present study. It is also possible that the initial (4 h) stimulation of BGT1 membrane insertion by NO may be followed by downregulation of transcription at 24 h. This could account in part for the fact that BGT1 upregulation by hypertonicity reaches a maximum after 24 h [28]. In conclusion, NO is another example of a rapidly acting regulator of the BGT1 transporter but, in contrast to other rapidly acting factors such as calcium, ATP and adenosine that downregulate BGT1 transport [12], NO upregulates the transporter and is not specific for BGT1 since SNAT2 is also upregulated. This supports the concept that one of the roles of NO produced by renal medullary cells during hypertonic stress [21] may be acceleration of the adaptive response of specific osmolyte transporters.
ACKNOWLEDGEMENTS
- Supported in part by a grant from the American Heart Association Midwest Affiliate (SAK), and by the Life-Health Science Internship at IUPUI (BMA). TIRF-FRAP studies were conducted in the Advanced Microscopy Core Facility at the University of Colorado Denver. We thank Dr HG Bohlen (Indiana University School, of Medicine) for helpful discusions.