-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Biophysics
p-ISSN: 2168-4979 e-ISSN: 2168-4987
2018; 8(1): 9-15
doi:10.5923/j.biophysics.20180801.02

Synthesis, X-Ray Structure and Molecular Docking Analysis of Two Novel 1,2,4,5-Tetrasubstituted Imidazole Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRatika Sharma 1, A. Jayashree 2, B. Narayana 2, B. K. Sarojini 3, C. Ravikumar 4, S. Murugavel 5, Sumati Anthal 1, Rajni Kant 1
1X-ray Crystallography Laboratory, Department of Physics, University of Jammu, Jammu Tawi, India
2Department of Studies in Chemistry, Mangalore University, Mangalagangotri, India
3Department of Studies in Chemistry-Industrial Chemistry Section, Mangalore University, Mangalagangotri, India
4Thanthai Periyar EVR Government Polytechnic College, Vellore, Tamilnadu, India
5Department of Physics, Thanthai Periyar Government Institute of Technology, Vellore, Tamilnadu, India
Correspondence to: Rajni Kant , X-ray Crystallography Laboratory, Department of Physics, University of Jammu, Jammu Tawi, India.
| Email: |  |
Copyright © 2018 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Imidazole derivatives I [1-(3,5-Difluoro-4-methylphenyl)-2-(4-fluorophenyl)-4,5-diphenyl-1H-imidazole and II 2-(4-fluorophenyl)-4,5-diphenyl-1-(4-trifluoro)-phenyl-1H-imidazole] have been synthesized by using multicomponent reaction(MCR). The compounds (I & II) are crystallized in the monoclinic space group I2/a and in triclinic space group P-1 with unit cell parameters a = 22.1693(7), b= 8.1636(6), c=25.7250(19) Å, β = 112.526(9)° and a = 9.2427(6), b = 13.4381(9), c =19.7520(4) Å, α= 90.464(5)°, β = 99.530(5)°, γ=106.210(6)˚ respectively. The complexes have been characterized by single crystal X-ray crystallographic analysis which revealed the asymmetric unit of the compound II contains two crystallographically independent molecules with almost similar geometries. The crystal structures were solved by Direct Methods and refined by Full Matrix least squares procedure. The C-H …F [inter-molecular and intra-molecular] interactions plays an important role in stabilizing the crystal packing. Molecular docking studies were used to identify the inhibitory activity against fungal protein cyctochrome P450 14 α demethylase enzyme.
Keywords: Imidazole, Crystal structure, Direct method, Intermolecular and intramolecular interactions, X-ray analysis, Molecular docking
Cite this paper: Ratika Sharma , A. Jayashree , B. Narayana , B. K. Sarojini , C. Ravikumar , S. Murugavel , Sumati Anthal , Rajni Kant , Synthesis, X-Ray Structure and Molecular Docking Analysis of Two Novel 1,2,4,5-Tetrasubstituted Imidazole Derivatives, International Journal of Biophysics , Vol. 8 No. 1, 2018, pp. 9-15. doi: 10.5923/j.biophysics.20180801.02.
Article Outline
1. Introduction
- Synthesis of multisubstituted imidazoles through multi-component reactions has got much attraction since they are an important class of pharmaceutical compounds and attractive targets in medicinal chemistry. They exhibit a wide spectrum of biological activities such as anti-inflammatory [1], antibacterial [2], antitumor, anti-diabetic [3]. The incorporation of the imidazole nucleus is an important synthetic strategy in drug discovery and finds tremendous applications in the treatment of several diseases [4]. It has been reported that these derivatives show various pharmacological activities such as analgesic [5], anti-depressants [6], antiviral [7], anticancer [8], anti-leishmanial [9] etc. In view of the numerous biological, pharmacological and material properties associated with 1,2,4,5-tetra substituted imidazoles, we report the synthesis and crystal structure of two novel 1,2,4,5-tetrasubstituted imidazole derivatives along with their docking studies. Infections caused by fungus are one of the major health problem faced by human beings presently. The occurrence of primary and adaptable nature of fungal infections persists to increase precipitously because of the increased number of immune compromised patients who are affected with tuberculosis, AIDS, cancer and organ transplantation [10-12]. Universal antifungal agents can be normally grouped based on their action mechanism in pathogenic fungi, primarily in cell membrane, cell wall and intracellular action [13]. Hence, constant search for antifungal agents on new targets or new mechanism of action is essential.Drug discovery programs are oriented to the search for lead structures. Virtual screening and molecular docking constitute great alternatives to find hit compounds. Azoles are the chief choice of drugs for the treatment of fungal infection high therapeutic index [14]. These facts directed us to quest for novel azoles lead compounds with more structural specificity for inhibit fungal enzymes. In this paper, molecular docking studies were carried out against the fungal protein cytochrome P450 sterol 14DM CYP-51 of C. albicans (PDB Id: 1E9X) [15] with the synthesized imidazole derivatives. The binding energy and interactions were found using AutoDock Vina [16]. The docking results were compared with the standard drug, clotrimazole, an antifungal medication which is sold in the brand name canesten to treat a wide variety of fungal infections like vaginal yeast infections, diaper rash, oral thrush, etc.
2. Experimental
2.1. Synthesis and Mechanism
- A mixture of benzil (1 mmol), substituted amine (1 mmol), aryl aldehyde (1 mmol), ammonium acetate (1 mmol) and ZnO nanoparticles (0.1 mmol) in glacial acetic acid was heated at 60°C under stirring for around 2 hrs. The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was cooled to room temperature. The reaction mixture was then poured into cold water. The solid separated was filtered by suction to afford crude product. The pure tetra-substituted imidazoles were obtained by further recrystallization from ethanol/water. Crystal of the purified products was generated in acetone/water. The complete reaction scheme is presented in Figure 1.
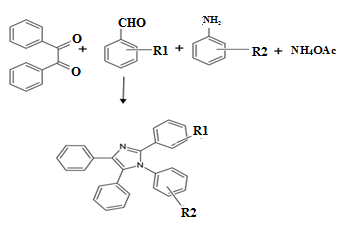 | Figure 1. Reaction scheme |
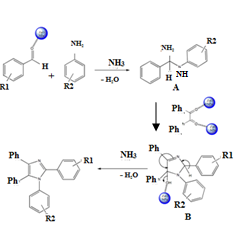 | Figure 2. A plausible mechanism for the formation of tetrasubstituted imidazoles |
2.2. X-ray Data Collection and Structure Refinement
- X-ray intensity data of both the compounds I and II (size; 0.30 X 0.20 X 0.10 mm) having well-defined crystal morphology were collected at 293(2) K on X’calibur CCD area-detector X-ray Diffractometer [17] equipped with MoKα radiation (λ=0.71073 Å). The cell dimensions were determined by the least-squares fit of angular settings of 1810(I), and 3330(II) reflections in the 𝜃 range 3.7 to 26.7°(I) and 4.1 to 26.0°(II), respectively. I > 2σ(I) criterion were employed to the unique data sets. Data were corrected for Lorentz and Polarization factors. The structures were solved by direct methods using SHELXS97 [18]. All non-hydrogen atoms of the molecules of both the structures were located from the best E-map and Full-matrix least-squares refinement was carried out using SHELXL97 [18]. The hydrogen atoms were geometrically fixed and allowed to ride on the corresponding non-H atoms. Atomic scattering factors were taken from International Tables for X-ray Crystallography (1992, Vol. C, Tables 4.2.6.8 and 6.1.1.4). CCDC 1484506 and 1479887 contains the supplementary crystallographic data for I and II respectively. The crystallographic data are summarized in Table 1.
|
|
|
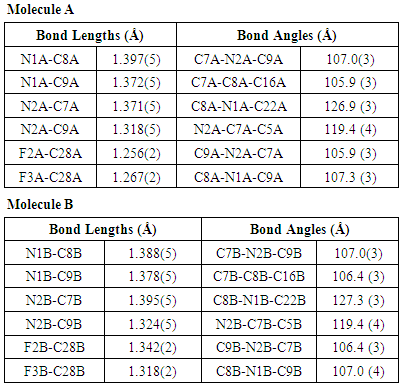
 | Figure 3. ORTEP view of the compound I with displacement ellipsoids at the 40% probability level. H atoms shown as small spheres |
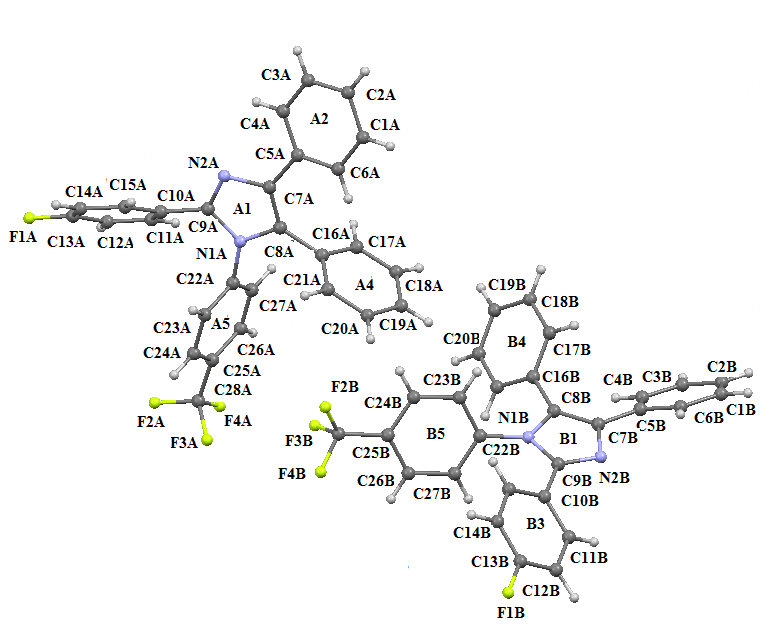 | Figure 4. ORTEP view of compound II with displacement ellipsoids at the 40% probability level. H atoms shown as small spheres of arbitary radii |
2.3. Molecular Docking Studies
- Molecular docking provides useful facts about drug receptor interactions and is frequently used to compute the binding orientation of small molecule drug candidates to their protein targets in order to calculate the affinity and activity of the small molecule. In our study, molecular docking is carried out using AutoDock Vina to find out the binding energy and interactions of synthesized imidazole derivatives with the protein, cytochrome P450 sterol 14DM CYP-51 of C. albicans (PDB Id: 1E9X) downloaded from Protein Data Bank (http://www.rcsb.org). The removing of hydrogen atoms and crystallized ligand structures of the protein along with addition of polar hydrogens were carried out using AutoDock Tools and the same were saved as PDBQT file. Ligand was also taken and saved in PDBQT file using ADT. The binding centre values based on active site present in protein with grid center coordinates (X= -17.143, Y= -6.199, Z= 64.493) and grid box size (x = 20, y = 20, z = 20). The docking results were viewed with Discovery Studio, 2015 [22].
3. Results and Discussion
3.1. 1-(3,5-Difluoro-4-Methylphenyl)-2-(4-Fluorophenyl)-4,5-Diphenyl-1H-Imidazole(I)
- The X-ray analyses reveal that both the molecules belong to the same series of compounds with different substitutions at the two phenyl rings. In compound I all bond lengths and angles are normal [23] and comparable with those reported for related compounds [24, 25]. All the rings in the molecule are planar. The three phenyl rings bonded to the imidazole at the position C7, C9, C8 and N1 are inclined to the imidazole ring at the dihedral angle 48.48(1), 24.10(2), 27.26(3) and 70.95(3)° respectively. The architecture of the crystal structure is determined mainly by one intermolecular [C-H…F] and one intramolecular [C-H…F] hydrogen bonds. C28 acts as the hydrogen bond donar in both inter and intra hydrogen bonds. C28-H28C…F1 interactions results in the formation of chains along the b-axis [Figure 5]. These chains of molecules are packed together to form two-dimensional network running along ac- plane [Figure 6].
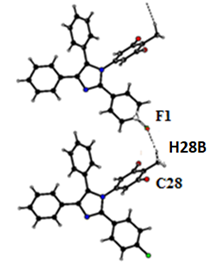 | Figure 5. Part of the crystal structure, showing the formation of chains. The dashed lines show intermolecular C-H…F hydrogen bond |
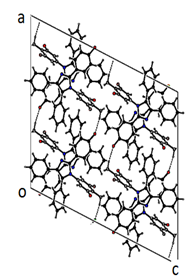 | Figure 6. The packing arrangement of compound I viewed in the ac-plane |
3.2. 2-(4-Fluorophenyl)-4,5-Diphenyl-1-(4-Trifluoro)-Phenyl-1H-Imidazole(II)
- The asymmetric unit of the compound II comprises of two crystallographically independent molecules, (A and B) with almost similar geometries. All the bond lengths and bond angles are normal [23-25] and in close agreement with each other except to those involving trifluromethyl group (F2/ F3/ F4/ C28) which shows significant variations in the two symmetry-independent molecules viz F1A-C28A= 1.263(8) Å; F1B-C28B= 1.342(5) Å, F3A-C28A= 1.211(8) Å; F3B-C28B= 1.318(5) Å & F4A-C28A=1.211(8); F4B-C28B=1.316(5) Å. The imidazole ring exhibits normal geometry [23] and is planar in both the asymmetric-independent molecules with maximum deviation for the atom C8A [0.0068 (5) Å] in molecule A and C7B [0.0032 (4) Å] in molecule B. The fluorine atom F1 has been found to lie in the plane of the atoms C10-C15 in both the molecules. The planarity of the fluorine atom has also been indicated by the value torsion angle F1A-C13A-C14A-C15A=179.7(2)° and F1B-C13B-C14B-C15B=179.2(2)°. The trifluromethyl ring is also coplanar with the plane of the ring defined by the atom C25 to C24. The architecture of the hydrogen bonding is determined mainly by two C-H…F and one C-H…N intra-molecular hydrogen bonds [Table 4]. The packing view of the molecules in compound II in the bc-plane is presented in the Figure 7.
|
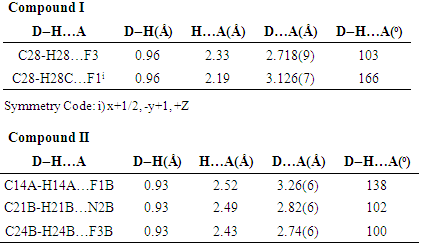
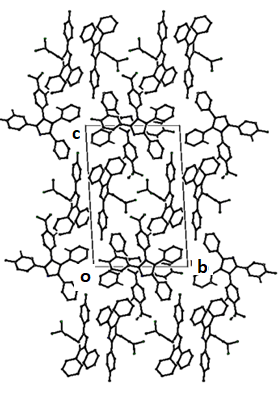 | Figure 7. Packing view of compound II |
3.3. Molecular Docking Analysis (I & II)
- Literature survey reveals that azole derivatives are good antifungal agents and they show good antifungal activity against cytochrome P450 sterol 14DM CPY51 of C. albicans, which is the best target to analyze the antifungal activity of the synthesized imidazole derivatives. The imidazole derivatives compounds (I) and (II) were docked with 14 α demethylase enzyme and was compared with the standard drug clotrimazole. Figure 8 shows the ligand-protein binding interactions of compound (I), compound (II) and Clotrimazole to 1E9X binding site. Table 5 shows the docked interactions and binding energy scores of compound (I), compound (II) and clotrimazole with the binding site of the target protein 14α-demethylase. The docking studies of synthesized compounds with 1E9X protein showed that the docking energy of -10.9 kcal/mol for compound(I)-1E9X complex and -11.3 kcal/mol for Compound (II)-1E9X complex, which are higher than that of standard drug clotrimazole-1E9X complex (-9.0 kcal/mol). The compound (I)-1E9X complex is stabilized by two hydrogen bonds, one halogen bond and one hydrophobic interaction. The residues LYS97 and HIS101 interact with the atoms F2 and F3 of the ligand, respectively, through hydrogen bond interactions. Also, a peculiar halogen bond is observed between the fluorine atom F1 and the residue ALA73. Further, a hydrophobic π-π interaction is observed between imidazole ring of the ligand and a six-membered ring of the residue TYR76.The compound (II)-1E9X complex is stabilized by two hydrogen bonds, one halogen bond and one electrostatic interaction. The residues LYS97 and ALA73 interact with the atoms F3 and F2 of the ligand, respectively, through hydrogen bond interactions. Also, a halogen bond is observed between the fluorine atom F2 and the residue GLN72. Further, an intermolecular cation-π interaction is observed between HZ3 atom of the residue LYS97 and trifluoro ring of the ligand. The Clotrimazole-1E9X complex is stabilized by one hydrogen bond with residue GLN72 and one hydrophobic sigma-π interaction with residue ALA256. As we know that, the occurrence of more number of interactions in the docking complexes will enhance the binding affinity and bioactivity of the compounds. As evident from Table 5, our synthesized compounds, show more interactions with the target protein 1E9X and also possess high docking energy score than the standard drug. Hence, the compounds I and II may be treated as vital inhibitors of 14α demethylase enzyme and as a drug for fungal diseases.
|
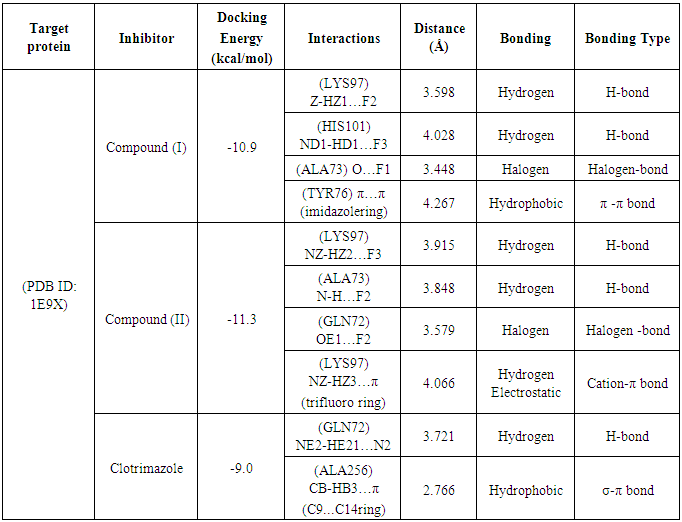
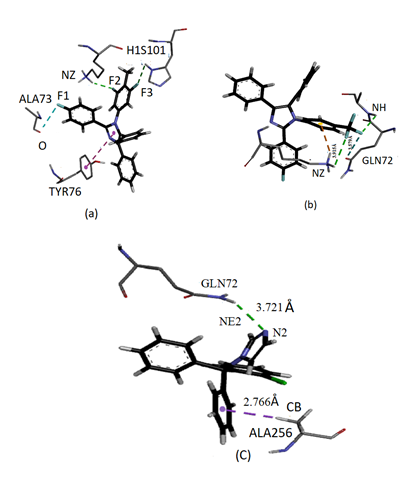 | Figure 8. Ligand-protein binding interactions of compounds (I), (II) and standard drug lotrimazole to 1E9 |
4. Conclusions
- Imidazole derivatives have occupied a unique place in the field of medicinal chemistry and is inclusion forms an important synthetic strategy in drug discovery. They are currently the most widely used insecticides in the world and are widely used for pest control in agriculture. Because of the wide application of these compound three biologically active Tetra-substituted imidazole derivatives (I & II) were synthesized by the multi-component reaction which provide us most powerful platform to access diversity as well as complexity in a limited number of reaction steps. The molecular structures have been determined by using X-ray diffractometer. The result of the single-crystal X-ray structure analysis establishes the structural aspects of these compounds. None of the structure exhibits any significant intermolecular interaction. In compound I the molecular packing is stabilized by inter molecular hydrogen bond. The packing of the other compound has been established by the presence of intra-molecular hydrogen bonds and van der waals forces.
ACKNOWLEDGEMENTS
- One of the authors (Rajni Kant) acknowledges the Indian Council of Medical Research (Research Project No: BIC/12(14)/2012) and the Department of Science and Technology (Research Project No EMR/2014/000467) for the financial support.