-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2019; 9(1): 11-21
doi:10.5923/j.bioinformatics.20190901.02

Comprehensive Computational Analysis Revealed Thirteen Novel Mutations in Human FSH-B gene Related to PCOS
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLNidal Essa 1, Enas A. Osman 2, Hadeel M. Yousif 2, Kutuf A. Albushra 2, Amel Nasir Eltayeb Ali 2, Tebyan Ameer Abdelhameed Abbas 2, Mohamed A. Hassan 2, 3
1Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology, Khartoum, Sudan
2Department of Bioinformatics, Africa city of Technology, Khartoum, Sudan
3Department of Bioinformatics, DETAGEN Genetic Diagnostics Center, Kayseri, Turkey
Correspondence to: Nidal Essa , Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology, Khartoum, Sudan.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background: Follicular stimulating hormone beta subunit (FSH-B) as gonadotropin hormone secreted from the anterior pituitary belongs to the glycoprotein family located on 11p14.1 and consists of three exons. It is responsible for follicular growth and ovarian steroidogenesis in females and spermatogenesis in males. PCOS is a common endocrinopathy affecting 4-20% of women within the reproductive age the pathophysiological process is not fully understood and lowering of serum FSH level due to gene polymorphism lead to abnormal folliculogenesis and irregular menstrual cycle. In this study, we used various computational approaches to identify nsSNPs which probably be deleterious to the structure and/or function of FSH-B protein that might be associated with polycystic ovary syndrome. Methods: The data on human FSH-B gene was retrieved from dbSNP/NCBI. Eleven different bioinformatics prediction algorithms; SIFT, Polyphen, PROVEAN, SNAP2, Pmut, PhD-SNP, I-Mutant and Project Hope were used to analyze the effect of nsSNPs on functions and structure of the FSH-B protein, and RaptorX for protein modeling and Chimera for visualization of the model, in addition, we used PolymiRTS to detect SNPs on miRNA binding sites. Results: After retrieval of SNPs from the NCBI database, 164SNPs were classified as missense SNPs. From functional analysis softwares, 84 SNP were predicted to be deleterious then they analyzed by disease - related software 13 SNPs, when checked for protein stability, 12 of them decreased protein stability and one SNP increased its stability. Conclusion: Consideration should be taken to these 13 novel mutations when we are carrying out genetic studies through human samples.
Keywords: PCOS, In Silico analysis, FSH-B, Folliculogenesis, Computational analysis
Cite this paper: Nidal Essa , Enas A. Osman , Hadeel M. Yousif , Kutuf A. Albushra , Amel Nasir Eltayeb Ali , Tebyan Ameer Abdelhameed Abbas , Mohamed A. Hassan , Comprehensive Computational Analysis Revealed Thirteen Novel Mutations in Human FSH-B gene Related to PCOS, American Journal of Bioinformatics Research, Vol. 9 No. 1, 2019, pp. 11-21. doi: 10.5923/j.bioinformatics.20190901.02.
Article Outline
1. Introduction
- The anterior pituitary glycoprotein hormone family consists of follicle-stimulating hormone, luteinizing hormone, chorionic gonadotropin, and thyroid-stimulating hormone [1]. All of these glycoproteins consist of an identical alpha subunit and a hormone-specific beta subunit. Follicular stimulating hormone beta subunit (FSH-B), also known as Follitropin subunit beta, located on 11p14.1 and consists of 3 exons, two transcript variants encoded for one protein with 129 amino acids which enables ovarian folliculogenesis to the antral follicle stage as well as Sertoli cell proliferation and maintenance of sperm quality in testis through binding to Follicular stimulating hormone receptor (FSHR) that expresses in the ovarian granulosa cells and Sertoli cells of the testis [1-3].Polycystic ovary syndrome (PCOS), the disease of our interest, is a complex disorder, in which both environmental and genetic factors contribute to its pathogenesis [4]. Despite extensive researches, the precise pathophysiology of PCOS remains unknown. New theories were introduced, and an interest in the genetics of PCOS has increased considerably during recent years [5-6].The etiology of PCOS is not fully understood, but the role of genetic factors has long been established by familial aggregation and genome-wide association studies revealing that altered expression of several genes affect signal transduction pathways controlling steroidogenesis, steroid hormones action, gonadotropin action and regulation, insulin action and secretion, energy homeostasis and chronic inflammation [5-6]. Common phenotypic pictures of PCOS vary among different populations due to environmental, genetics and diagnostic criteria that define the appearance of the disease among women with PCOS such as irregular menstrual cycles, hyperandrogenism, polycystic ovarian morphology and acne [6-9].In particular, Hypothalamus pituitary gonadal axis (HPO) genes as reproductive genes responsible for the production of steroid hormones and ovulatory process, any defect in the gene or its receptor due to mutations will result in structure and biological dysfunction which consequently results in anovulation and irregular menstrual cycle [10]. FSHB is thus likely to play an important role in the etiology of PCOS. Missense variants as one of these mutations lead to altering the translation of amino acid which consequently results in protein dysfunction either by their effect on protein solubility or by disrupting protein structure. Recent genome-wide association studies (GWAS) in Han Chinese women with PCOS demonstrate 11 genetic loci that associated with PCOS which are responsible for steroidogenesis, steroid hormones action, gonadotropin action and regulation, insulin action and secretion, energy homeostasis, chronic inflammation, and others. Replication studies have demonstrated that variants at several of these loci also confer risk for PCOS in women of European ethnicity. The strongest loci in Europeans contain genes for DENND1A and THADA, with additional associations in loci containing the LHCGR, FSHR, YAP1, and RAB5/SUOX. [4,7,11].PCOS is a very complex syndrome, with a lot of undiscoverable genetic components that associate with its pathogenesis. Abnormal folliculogenesis that associated with PCOS is due to low serum FSH level but its mechanism of occurrence is unknown. Generally, these mutations affect its expression on the ovarian granulosa and result in abnormal serum level. Reported FSHB variants (rs11031010, rs11031006) showed significant association with abnormal serum levels of FSH and LH at genome-wide significance among both Chinese and European PCOS populations [12-16].Exploitation of Bioinformatics algorithms with the construction of databases enables biomedical scientists to collect, retrieval, and analyze scientific data on a mass scale [17-18].Combination of bioinformatics resources and computational ''In-Silico'' experiment will accelerate the pace and efficiency of scientific discovery [19-21].This is the first study used the in silico analysis for building up knowledge about PCOS. Thus, we aimed to determine the influence of various SNPs in FSH-B gene using in silico prediction software and assessing their effects on the structure/function of FSH protein that may have an important role in disease susceptibility which will be examined through wet laboratory work and help in understanding the genetic mechanisms of the disease through these useful tools.
2. Material and Methods
- 2.1. Data retrieval: from the dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), the SNPs information regarding FSH-B gene was obtained (SNP ID), the protein sequence and it is accession number was collected from Uni Prot database. http://www.uniprot.org.2.2. SIFT (Sorting Intolerant from Tolerant): https://sift.bii.a-star.edu.sg/. As functional analysis tool based on the two concepts, sequence homology and conservation regions on protein sequence, the SFIT server predicts which amino acid substation lead to deleterious effect (SIFT scores <0.05) or tolerated effect (SIFT scores >0.05) on the protein structure [17]. 2.3. Polyphen-2 (Polymorphism Phenotyping v2), http://genetics.bwh.harvard.edu/pph2/: Based on the position-specific independent count scores (PSIC) which calculated in dependence of the multiple sequence alignment analysis and protein 3D structure of the wild and the mutant protein. Its outcomes values ranged from (0-1) classified the predication product to benign, possibly damaging and probably damaging. MsSNPs that predicted to be intolerant by SIFT has been submitted to Polyphen as protein sequence in FASTA format that obtained from UniproktB /Expasy after submitting the relevant ensemble protein (ESNP) there, and then we entered the position of mutation, native amino acid and the new substituent for both structural and functional protein analysis [18].2.4. Provean (Protein Variation Effect Analysis) (http://provean.jcvi.org/index.php): It is a software tool that predicts the impact of amino acid substitution on the biological function of the protein when we entered the protein FASTA sequence along with amino acid substitutions as input query for analysis. The outcome product according to the threshold score of −2.5 as tolerated or deleterious effect [19].2.5. SNAP2 (Screening of Non-Acceptable Polymorphism 2; https://rostlab.org/services/snap2web/): It is a neural network dependent software tool that distinguishes between effect and neutral variants/non-synonymous SNPs by taking a variety of sequence and variant features into account. After entering the protein FASTA sequence as input query while the output predication as neutral, non- neutral effect [20].2.6. PhD-SNP (Predictor of human Deleterious SingleNucleotide Polymorphisms; http://snps.biofold.org/phd-snp/phd-snp.html): It is an online Support Vector Machine (SVM) based classifier, is optimized to predict if a given single point protein mutation can be classified as disease-related or as a neutral polymorphism, also we used the protein FASTA sequence as input query besides the residues change to obtain the output results [21].2.7. SNPs &GO (Single nucleotide polymorphism & Gene Ontology, PHD-SNP (http://snps.biofold.org/snps-and-go). It's an accurate algorithm method that used the FASTA sequences of protein to give unique framework information evolutionary information and function as encoded in the Gene Ontology terms, and other outperforms with available predictive methods [22].2.8. P-mut (http://mmb2.pcb.ub.es:8080/PMut): a web-based tool for the annotation of pathological variants on proteins based on the uses of neural networks. The output display according to the pathogenicity index ranged from 0-1; more than 0.5 scored as single pathological mutations [23].2.9. I–Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi): In regard to the neural network concept, it predicts the changes in protein stability in response to the occurrence of mutation. The output obtained is either increased or decreased stability of the protein [24]. 2.10. Project Hope (http://www.cmbi.ru.nl/hope/): It is a new online web-server to search protein 3D structures (if available) by collecting structural information from a series of sources, including calculations on the 3D coordinates of the protein, sequence annotations from the Uni Prot database, and predictions by DAS services. Submission of protein sequence and the mutant variants as input information to the web server which displays the output as textual report supported by figures and animations [25].2.11. RaptorX (http://raptorx.uchicago.edu/): It is a web server predicting structure property of a protein sequence without using any templates. It outperforms other servers, especially for proteins without close homologs in PDB or with very sparse sequence profile. The server predicts tertiary structure [26].2.12. PolymiRTS Database 3.0: As well as it is a database of naturally occurring DNA variations in microRNAs (miRNA) seed region and miRNA target sites, it is used to predict 3 un-translated region polymorphism (3’ UTR) effect upon microRNAs and their target sites which may influence miRNA-mRNA interaction, causing impact on miRNA-mediated gene repression. PolymiRTS database was made by examining 3UTRs of mRNAs in human and mouse for SNPs in miRNA target destinations. Then, the impact of polymorphism on gene expression and phenotypes are identified and then connected in the database. The PolymiRTS data base also includes polymorphism in target sites that have been supported by a variety of experimental methods and polymorphism in miRNA seed regions [27].2.13. GeneMANIA: (http://www.genemania.org/). It is a web interface that predicts gene interaction with other genes in various pathways using genetic interactions, pathways, co-expression, co-localization and protein domain similarity [28].
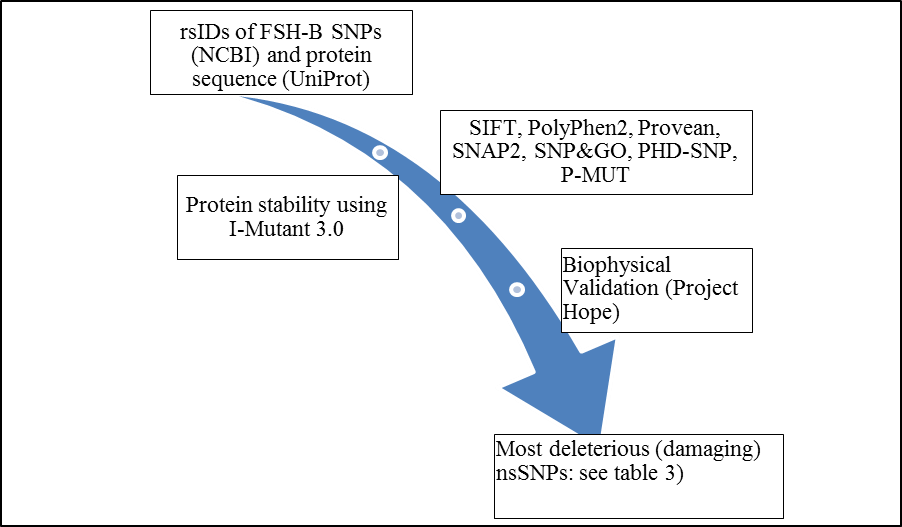 | Figure 1. Diagrammatic representation of FSH-B gene in silico work flow |
3. Results
- Using dbSNP database for gene data set, the human FSH-B gene, gene of our interest, contained a total of 84 SNPs missense nsSNPs, 13 nsSNPs of them were predicted to have the most damaging effects on FSH-B protein's structure and function.
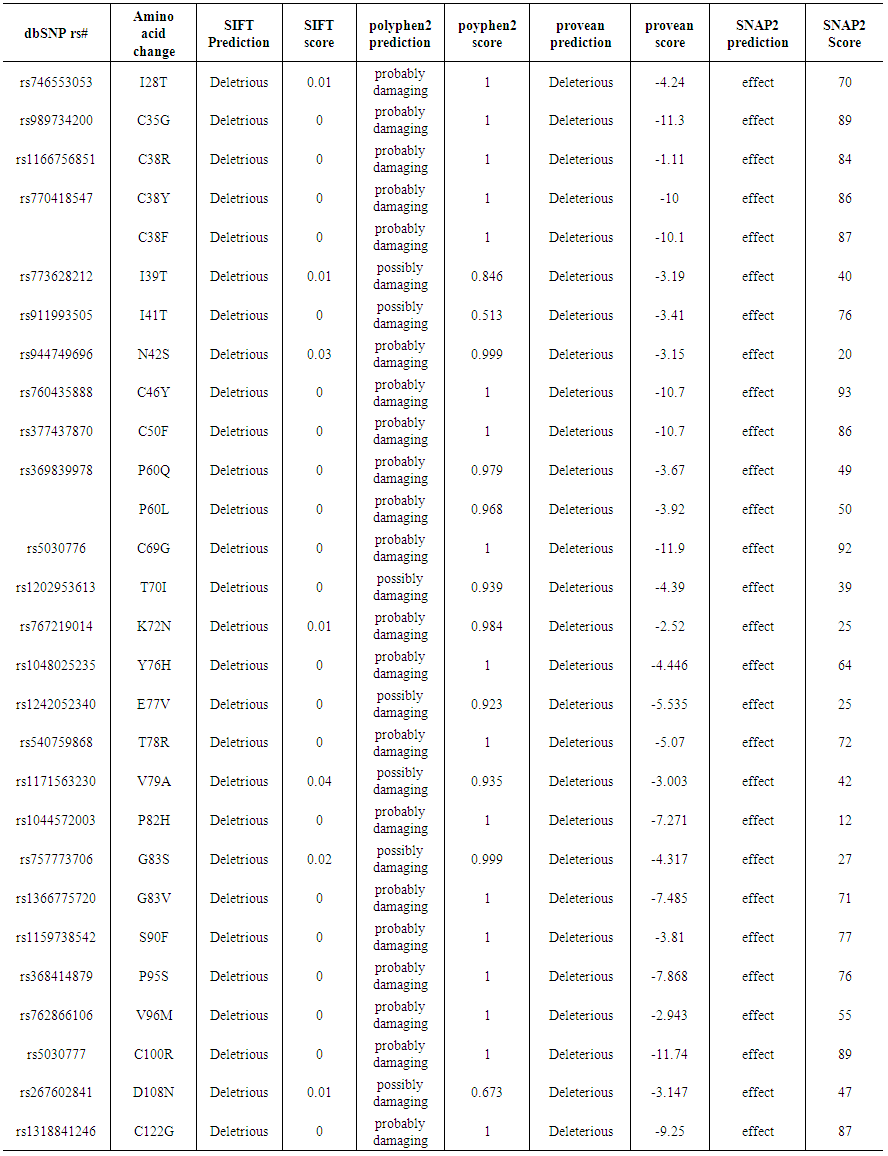 | Table (1). Damaging or Deleterious or effect nsSNPs associated variations predicted by SIFT, Polyphen, PROVEAN, SNAP2 softwares |
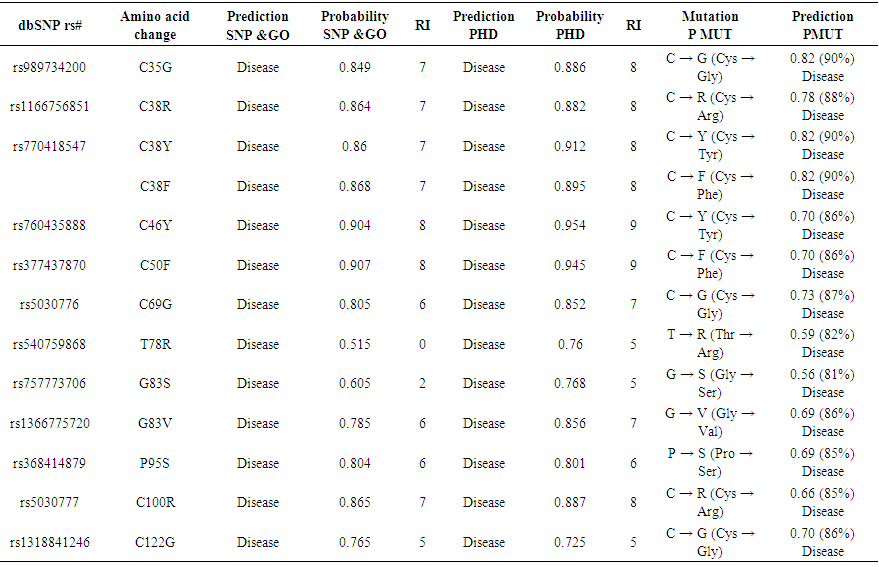 | Table (2). Disease effect nsSNPs associated variations predicted by Pmut, SNP&GO and PHD softwares |
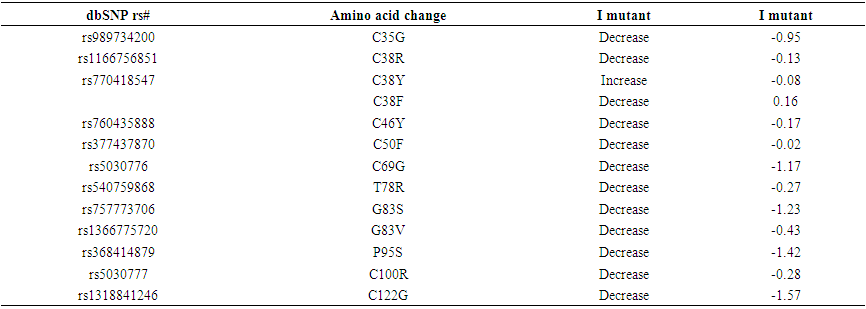 | Table (3). Stability analysis predicted by I-Mutant version 3.0 (also Show the novel mutations) |
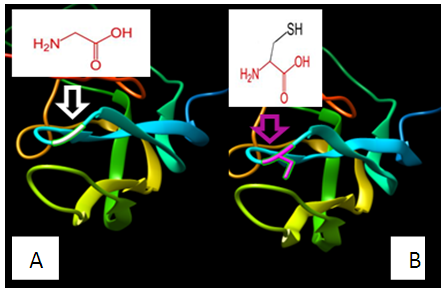 | Figure 2. SNP ID: rs989734200, (C35G) The amino acid Cysteine in the native protein (A) changed to Glycine in the mutant protein (B) at position 35 |
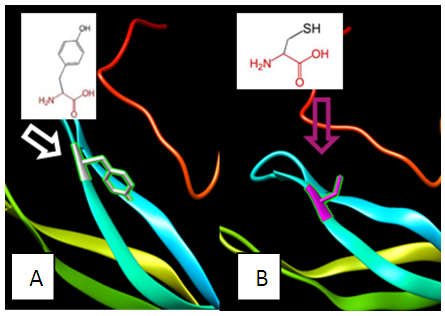 | Figure 3. SNP ID: rs770418547, (C38F) The amino acid Cysteine in the native protein (A) changed to Phenylalanine in the mutant protein (B) at position 38 |
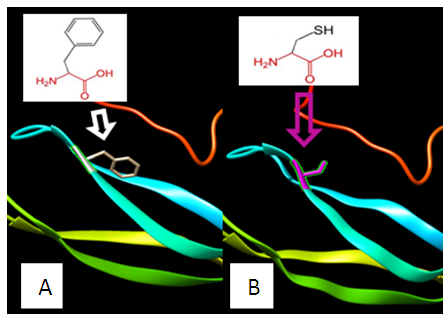 | Figure 4. SNP ID: rs770418547, (C38R) The amino acid Cysteine in the native protein (A) changed to Tyrosine in the mutant protein (B) at position 38 |
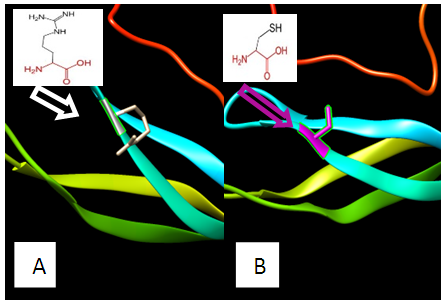 | Figure 5. SNP ID: rs1166756851, (C38R) The amino acid Cysteine in the native protein (A) changed to Arginine in the mutant protein (B) at position 38 |
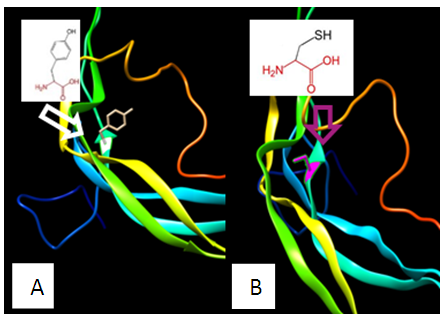 | Figure 6. SNP ID: rs760435888, (C46Y) The amino acid Cysteine in the native protein (A) changed to Arginine in the mutant protein (B) at position 46 |
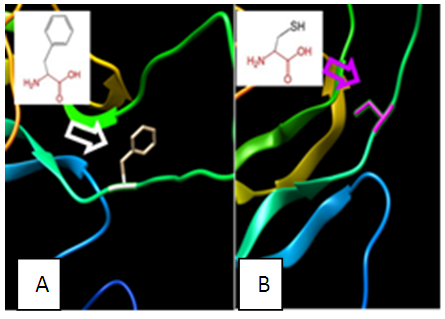 | Figure 7. SNP ID: rs377437870, (C50F) The amino acid Cysteine in the native protein (A) changed to Phenylalanine in the mutant protein (B) at position 50 |
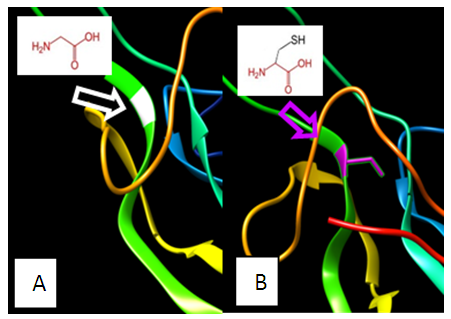 | Figure 8. SNP ID: rs5030776, (C69G) The amino acid Cysteine in the native protein (A) changed to Glycine in the mutant protein (B) at position 69 |
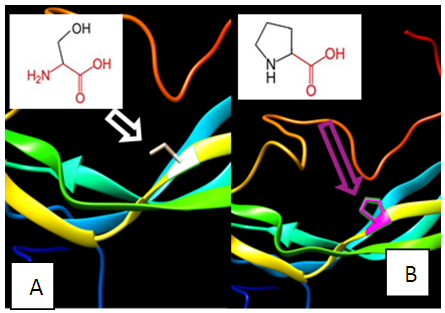 | Figure 9. SNP ID: rs368414879, (P59S) The amino acid Proline in the native protein (A) changed to Serine in the mutant protein (B) at position 59 |
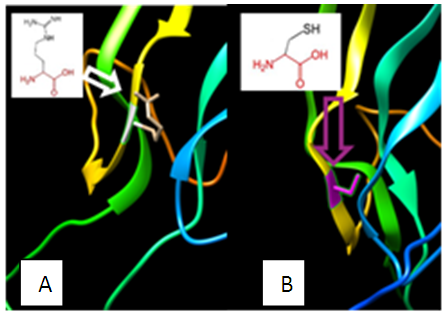 | Figure 10. SNP ID: rs5030777, (C100R) The amino acid Cysteine in the native protein (A) changed to Arginine in the mutant protein (B) at position 100 |
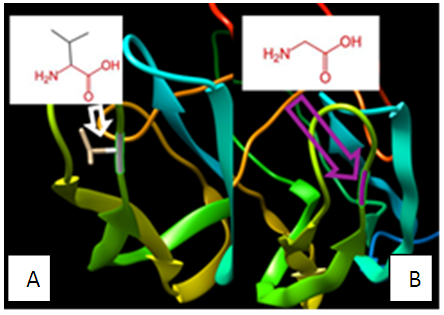 | Figure 11. SNP ID: rs1366775720, (G83V) The amino acid Glycine in the native protein (A) changed to Valine in the mutant protein (B) at position 83 |
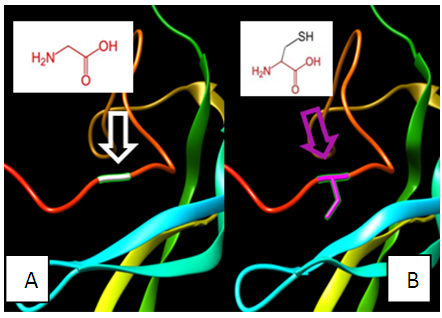 | Figure 12. SNP ID: rs1318841246, (C122G) The amino acid Cysteine in the native protein (A) changed to Glycine in the mutant protein (B) at position 122 |
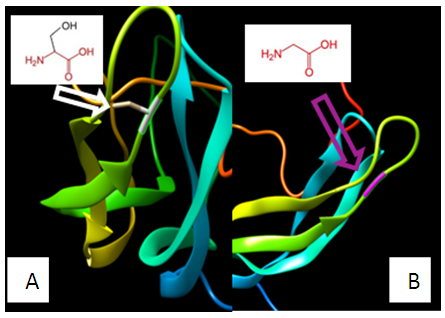 | Figure 13. SNP ID: rs757773706, (G83S) The amino acid Glycine in the native protein (A) changed to Serine in the mutant protein (B) at position 83 |
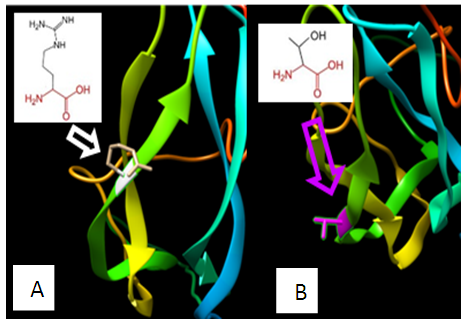 | Figure 14. SNP ID: rs540759868, (T78R) The amino acid Threonine in the native protein (A) changed to Arginine in the mutant protein (B) at position 78 |
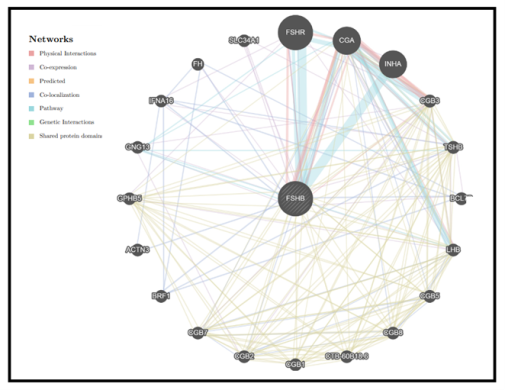 | Figure 15. GeneMANIA result for FSH-B Gene |
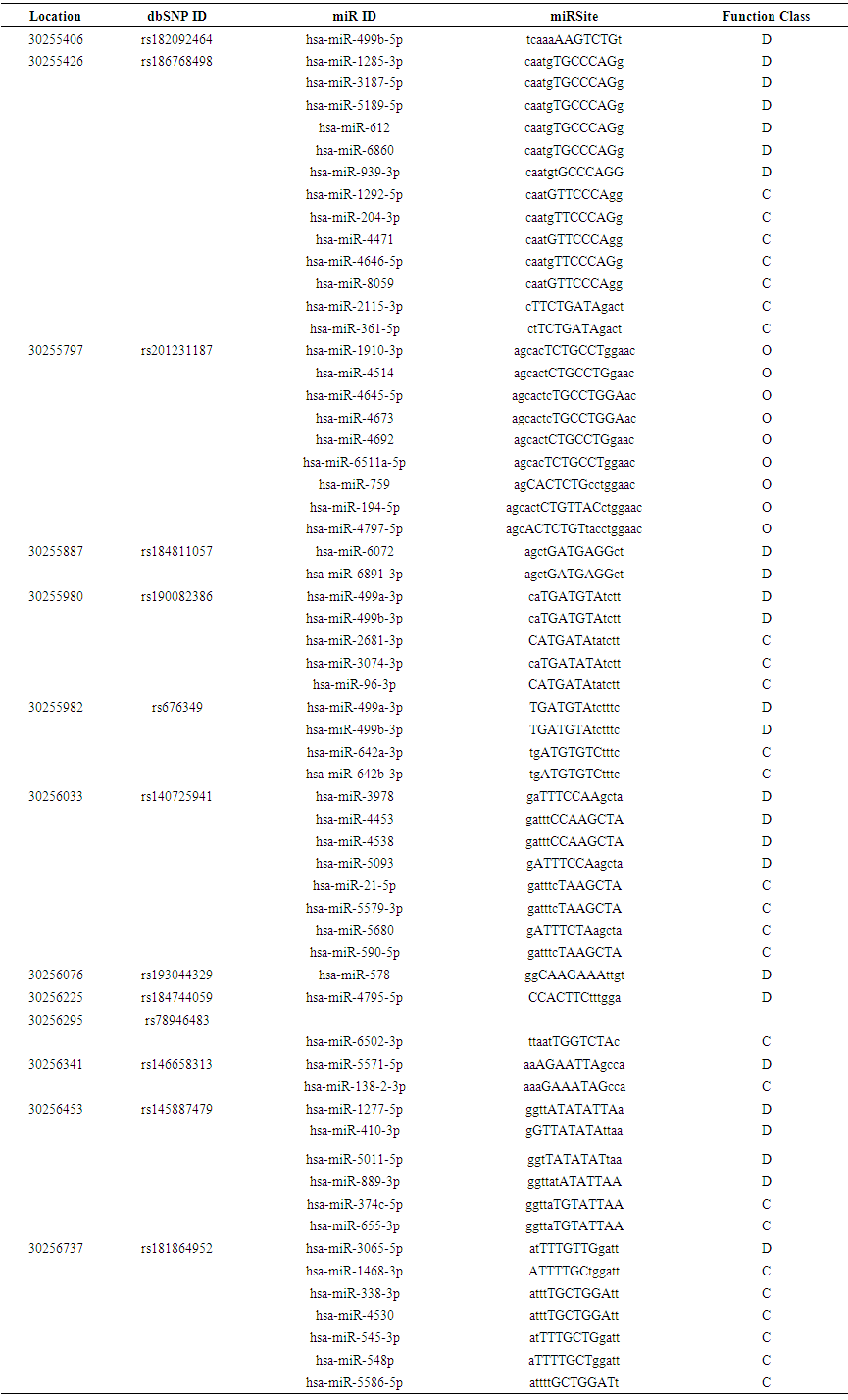 | Table (4). Prediction of SNPs at the 3’UTR Region using PolymiRTS |
4. Discussion
- FSHB gene was investigated in dbSNP National Centre of Biotechnology Information (NCBI public database). This gene contains a total of 354 SNPs in coding region; of which 76 are missense, 4 synonymous, eight nonsense, five frame shift and 612 are in the non-coding region, of which 300 in the 3'un-translated region (3’ UTR) and 312 in 5’ un-translated region (5’UTR). We selected the missense coding SNPs and 3′UTR SNPs for our investigation.13 novel mutations have been predicated which had an effect on protein stability and function using different bioinformatics algorisms such as SIFT, PolyPhen-2, Provean, SNAP2, SNP&GO, PHD-SNP, P-MUT and I-Mutant 3.0 (Figure 15).Changes in physiochemical properties of the protein due to mutations will possibly result in critical changes in the protein function and consequently result in different phenotypic pictures of the disease.Regarding protein stability, all of the predicted SNPs were predicted to deregulates the protein stability, (rs770418547, C38Y) which increased protein stability. While four of them, ((rs989734200, C35G), (rs5030776, C69G), (rs1318841246, C122G) and (rs368414879, P59S)), their substitutions result in lowering the protein size and lead to losing of hydrophobic interactions in the protein core, while the rest increase the molecule size which disturbs interactions with other molecules or other parts of the protein.Additionally, three of them ((rs760435888, C46Y), (rs377437870, C50F) and (rs5030777, C100)) were unique in their position and lead to complete damaging of the protein which associated with protein folding problems.In this mutation, (rs989734200, (C35G)); the mutant residue is smaller than the wild type residue which will cause an empty space in the core of the protein, loss of hydrophobic interactions property and decrease protein stability.Substitution of Cysteine to Phenylalanine and Arginine rs770418547, (C38F and C38R) in the same location 38 due to mutation has many effects in protein structure appear in decreasing protein stability, increasing the size which can disturb interactions with other molecules or other parts of the protein. While in the same location when Cytosine is substituted to Arginine (C38R) the protein become positive in charge, loss of hydrophobic interaction and disturbs its interactions with other molecules or other parts of the protein.Also mutation in this location rs1166756851, (C38R); results in increasing the size of the protein that changes the physiochemical property of the protein to a positive charge while the wild-type residue charge is neutral which consequently might result in losing hydrophobic interactions with other molecules on the surface of the protein.In this unique location rs760435888, (C46Y); Cytosine is also mutated to Tyrosine results in complete damaging of the protein, which might disturb the interactions with other molecules or other parts of the protein because this residue is located on the surface of the protein.Furthermore, changing of Cytosine to Phenylalanine in location 50 (rs377437870, (C50F)) consequently damage the protein structure, increasing the size of it and can disturb interactions with other molecules or other parts of the protein.One of the mutations that leads to decrease protein size occur when Cysteine is converted to Glycine (rs5030776, (C69G)) and cause a possible loss of external interactions beside loss of hydrophobic interactions with other molecules on the surface of the protein, As well as, when altering of Proline to Serine localized in 59 position (rs368414879, (P59S)), the mutant residue is smaller in size and possibly causes loss of external interactions and hydrophobic interactions with other molecules on the surface of the protein.Conversion of Cysteine to Arginine in position 100 due to mutation (rs5030777, (C100R)) changes protein neutrality to positive charge and increases the size which consequently damages it due to protein folding problems with loss of hydrophobic interactions in the core of the protein. rs1366775720, (G83V); showed that mutant residue is more hydrophobic than the wild-type residue, bigger in size, and the torsion angles of this residue are unusual. Only glycine is flexible enough to make these torsion angles, mutation into another residue will force the local backbone into an incorrect conformation and will disturb the local structure of the protein. rs1318841246, (C122G); the mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions and loss of hydrophobic interactions with other molecules on the surface of the protein.Moreover, conversion of Glycine to Serine in position 83 (rs757773706, (G83S)) associated with decrease protein stability, bigger size of the mutant residue and the unusual torsion angles will force the local backbone into an incorrect conformation and will disturb its local structure.Mutated Arginine in position 78 (rs540759868, (T78R)) changes the protein neutrality to a positive charge, become bigger in sizeand might cause loss of hydrophobic interactions with other molecules on the surface of the protein.Despite extensive genetic studies were done to study the association of FSH-B polymorphisms with PCOS, our computational analysis of the gene revealed that these thirteen novel SNPs success in the analysis by various softwares with no previous studies.Only two of them (rs5030776, rs5030777) were reported by Nagirnaja, L [29], et al., Lindstedt, G, et al [30], and Layman, L.C, [31] et al., associated with increase serum FSH level and male infertility. FSH-B plays the same role in development and maturation of gametes in both genders and regard to female physiology; it plays a central role in the regulation of ovarian folliculogenesis, which is disordered in PCOS, under the influence of LH action.FSH-B gene interacts with more than 20 genes to accomplish different biological functions. Therefore, damaging its protein due to one of these mutations will probably destroy these pathways.Consideration should be taken to these thirteen novel mutations when we are carrying out genetic studies through human samples.
5. Conclusions
- 13 nsSNPs with different positions were predicted to be the most damaging mutations for FSH protein, altering its physiochemical properties such as size, charge, hydrophobicity, and stability, leading to loss or disturbance of the protein internal and external interactions and eventually loss of the protein's function as well as disease association. Furthermore, the 61 SNPs at the 3UTR were predicted to disrupt miRNA binding sites and hence affect the gene expression functionCurrently, in silico analysis plays an essential role in narrowing the gap between the theoretical and applied parts of the medical research and by computational bioinformatics algorithms, we can increase our knowledge about disease pathogenic process which impacts in di covering newly preventive and treatment strategies tools.
ACKNOWLEDGEMENTS
- The authors desire to acknowledge the excited cooperation of Africa City of Technology-Khartoum, Sudan and to Dr. Mounkaila Noma for language editing and proofreading of the manuscript, University of Medical Sciences and Technology, Khartoum, Sudan.