-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2018; 8(1): 1-11
doi:10.5923/j.bioinformatics.20180801.01

Computational Analysis of Deleterious Single Nucleotide Polymorphisms (SNPs) in Human CALR Gene
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSahar G. Elbager1, Hadeel F. Gad Mahgoob2, Mohamed Y. Basher3, Asia M. Elrashid4, Hazem Abdo3, Shazalia K. Babiker1, Ali I. Alsaid5, Mohamed A. I. Alfaki6, Safinaz I. Khalil7, Amar A. Dowd1, Magdi A. Bayoumi1
1Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology (UMST), Sudan
2Faculty of Science, University of Princess Nourah bint Abdul Rahman, Saudi Arabia
3Faculty of Veterinary Medicine, University of Khartoum, Sudan
4Faculty of Science, University of Khartoum, Sudan
5Faculty of Science and Technology, Omdurman Islamic University, Sudan
6Faculty of Computer Science, Neelain University, Sudan
7Faculty of Medicine, University of Medical Sciences and Technology (UMST), Sudan
Correspondence to: Sahar G. Elbager, Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology (UMST), Sudan.
| Email: |  |
Copyright © 2018 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background: The human Calreticulin (CRT) is a multifunctional protein encoded by CALR gene (CALR) located on chromosome 19. Calreticulin plays an important role in protein folding and calcium homeostasis. It’s also has been associated with cell proliferation, cell cycle progression and immunogenic cell death. High CRT serum levels were reported in various cancer like prostate cancer and breast carcinoma. Being observed as an important molecule in biological responses and its association with various diseases, we aimed to systematically explore the probable effects of CALR genetic variants on functions and structure of calreticulin using in silico prediction softwares. Methods: The data on human CALR gene was retrieved from dbSNP/NCBI. Eleven different prediction algorithms; SIFT, Polyphen, PROVEAN, SNAP2, Condel, Pmut, nsSNPs Analyzer, PhD-SNP, I-Mutant, Mutpred and Project Hope were used to analyzing the effect of nsSNPs on functions and structure of the CRT protein. STRING and KEGG database were used for CRT protein-protein interaction. Results: As per dbSNP database, the human CALR gene investigated in this work contained a total of 682SNPs:53 SNPs in 3′ UTR region, 25 SNPs in 5′ UTR region, 343 SNPs in intron region, 103 SNPs in coding synonymous regions and 154 non-synonymous SNPs (nsSNPs) which comprises of 150 missense mutations, 3 frameshift mutations and one nonsense mutation. We selected missense nsSNPs for our investigation. A total of 4 nsSNPs P216L, R73C, W261G and Y128C were predicted to have the most damaging effects on CRT protein's structure and function. STRING and KEGG revealed that CRT protein had strong interactions with proteins that involved in protein processing and presentation networks. Therefore, any structural alterations in the CRT protein that interfere or harm these networks interactions would probably increase susceptibility to diseases. Conclusion: Based on these analyses, the present study suggested that the reported functional SNPs may act as potential targets in genetic association studies.
Keywords: CALR gene, Calreticulin, Computational analyses, Deleterious SNPs, Regulatory SNPs, Single nucleotide polymorphism (SNP)
Cite this paper: Sahar G. Elbager, Hadeel F. Gad Mahgoob, Mohamed Y. Basher, Asia M. Elrashid, Hazem Abdo, Shazalia K. Babiker, Ali I. Alsaid, Mohamed A. I. Alfaki, Safinaz I. Khalil, Amar A. Dowd, Magdi A. Bayoumi, Computational Analysis of Deleterious Single Nucleotide Polymorphisms (SNPs) in Human CALR Gene, American Journal of Bioinformatics Research, Vol. 8 No. 1, 2018, pp. 1-11. doi: 10.5923/j.bioinformatics.20180801.01.
Article Outline
1. Introduction
- The human Calreticulin (CRT) is a 46 KDa multifunctional protein predominantly located in endoplasmic reticulum (ER) [1] and it is also known as calregulin, CaBP3, CRP55, calsequestrin-like protein, and endoplasmic reticulum resident protein 60 (ERp60). It is encoded in humans by CALR gene (CALR) located on chromosome 19p13.13 [1].CRT molecule consists of three domains, including N-domain, P-domain, and C-domain. The N-terminal region of CRT is encoded by a highly conserved sequence that is folded in a stable globular domain containing eight antiparallel β-strands [2]. The N-terminal is an important functional domain that includes polypeptide and carbohydrate-binding sites, Zn2+ binding site and DNA-binding site of steroid receptor [3]. The disulfide bond formed by cysteine residues within this domain may interact with P-domain to produce important chaperone function of calreticulin [4]. The Proline-rich P-domain contains two sets of three repetitive amino acid sequence regions. These repeated amino acid sequences form the lectin-like chaperone structures which are responsible for protein-folding function of CRT [4]. In addition, the P-domain comprises a region with a high-affinity and low-capacity Ca2+-binding region [5]. The C-domain of CRT is a highly acidic region [4]. It binds to Ca2+ with high capacity and low affinity [4]. This terminal contains a KDEL sequence [lysine, aspartate, glutamate and leucine] ER retention motif which prevents CRT from being secreted from the ER [5]. These three domains are illustrated in Figure 1.
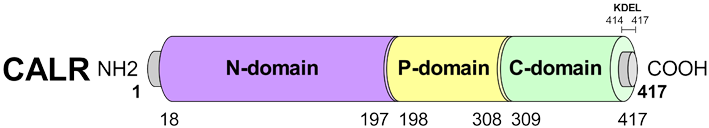 | Figure 1. Schematic primary structure of CALR protein. The conserved P-domain, N-domain and C-domain and KDEL motif are illustrated. Amino acid (aa) positions are indicated [6] |
2. Material and Method
2.1. Data Set
- The data on human CALR gene (accession ID: NP_004334) was retrieved from the Entrez Gene databases from National Center for Biological Information (NCBI) database on 13 March 2018. The CRT protein sequence (accession ID: P27797) and SNPs information of the CALR gene were obtained from UniProtKB databases (http://www.uniprot.org) and NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp/) respectively.
2.2. Identification of Deleterious nsSNPs
- To determine the functional impact (deleterious, damaging or natural), coding nsSNPs were analyzed using five different tools (SIFT, Polyphen -2, PROVEAN, SNAP2 and Condel). nsSNPs predicted to be deleterious by these five tools that were categorized as high-risk nsSNPs were subjected for further analysis like association with disease, stability analysis and structural effect using different tools. STRING and KEGG database were used for CRT protein-protein interaction.2.2.1 SIFT (Sorting intolerant from tolerant; http://siftdna.org/www/SIFT_dbSNP.html) server was used to identify the tolerated and deleterious SNPs. The effect of amino acid substitution on protein structure was assessed on the basis of degree of conservation of amino acids using sequence homology Substitution of an amino acid at each position with probability < 0.05 is predicted to be deleterious and intolerant, while probability ≥ 0.05 is considered as tolerant [27]. 2.2.2 Polyphen -2 (polymorphism and phenotype; http://genetics.bwh.harvard.edu/pph2/) server was used to predict the functional impact of amino acid substitution on protein structure and function based on sequence based characterization. The Prediction outcome was obtained in the form of probability score which classifies the variations as ‘probably damaging’, ‘possibly damaging’ and ‘benign’ [28]. UniProt protein accession ID P27797 along with position and name of wild type and variant amino acids of screened nsSNPs were submitted as query.2.2.3 PROVEAN (Protein Variation Effect Analyzer; http://provean.jcvi.org/index.php) is used to predict the possible impact of a substituted amino acid and indels on protein structure and biological function. It analyses the nsSNPs as deleterious or natural, if the final score was below the threshold score of −2.5 were considered deleterious; scores above this threshold were considered neutral [29]. The input query is a protein FASTA sequence along with amino acid substitutions.2.2.4 SNAP2 (Screening of Non-Acceptable Polymorphism 2; https://rostlab.org/services/snap2web/) is a tool, developed based on a neural network classification method which is freely available. It predicts the effect of nsSNPs on protein function [30].The input query submitted is the protein FASTA sequence and lists of mutants which provided scores of each substitution that can then be translated into binary predictions neutral or non-neutral effect natural.2.2.5 Condel (CONsensus DELeteriousness score of missense SNVs; http://bbglab.irbbarcelona.org/fannsdb/ query/condel). Condel is a method, used assess the outcome of non-synonymous SNVs, via applying a CONsensus DELeteriousness score that combines various tools. It computes a weighted approach of missense mutations from the complementary cumulative distributions of scores of deleterious and neutral mutations [28]. The input query submitted is rhe UniProt protein accession ID P27797 along with the position and name of wild type and variant amino acids of screened nsSNPs.
2.3. Prediction of Disease Associated SNPs
- The SNPs occurring in the protein coding region may lead to the deleterious consequences in its 3D structure and thus may lead to disease-associated phenomena. Here nsSNPAnalyzer, Pmut and PhD- SNP were used as tools to examine the disease-associated nsSNP occurring in the CALR protein coding region.2.3.1 nsSNPAnalyzer (http://snpanalyzer.uthsc.edu) is a tool, employed to predict the phenotypic effect (disease-associated vs. neutral) of a nsSNP by using a machine learning method called Random Forest, and extracting structural and evolutionary information from a query nsSNP [31]. The input was provided in form of protein FASTA sequence along with amino acid substitution.2.3.2 Pmut (http://mmb2.pcb.ub.es:8080/PMut) web server was used to predict disease-associated mutations. PMUT method is based on the use of neural networks (NNs) which is trained on large database of neutral mutations (NEMUs) and pathological mutations of mutational hot spots, which are obtained by alanine scanning, massive mutation, and genetically accessible mutations. The final output is displayed as a pathogenicity index ranging from 0 to 1 (indexes > 0.5 single pathological mutations) [32]. UniProt accession ID of CRT (P27797) along with name and position of wild type and mutant amino acid was submitted as an input for this server.2.3.3 PhD-SNP (Predictor of human Deleterious Single Nucleotide Polymorphisms; http://snps.biofold.org/phd-snp/ phd-snp.html) web server was used to predict if a given single point protein mutation can be classified as a disease-related or as neutral polymorphism. This server was mainly based on the support vector machines which can corroborate all the information regarding variations from the existing databases [33]. The input FASTA sequences of protein along with the residues change were submitted to PhD-SNP server for the analysis.
2.4. Prediction of nsSNPs Impact on the Protein Stability by I-Mutant2.0
- I-Mutant2.0 is a tool used for prediction of changes in protein stability due to single site mutations under different conditions (http://gpcr.biocomp.unibo.it/cgi/predictors/ I-Mutant2.0/I-Mutant2.0.cgi). It is a web server based on support vector machine which worked on dataset derived from Protherm, a database of experimental records on protein mutations. It can predict the stability changes in protein with 80% accuracy based on its structure and with 77% of accuracy based on its sequence [34]. The input can be submitted as either in the form of protein sequence or on a structure basis. For the present study, input was submitted in the form of protein FASTA sequence.Analyzing the effect of nsSNPs on physiochemical properties of the proteins.
2.5. Analyzing the Effect of nsSNPs on 3D Structure of the Proteins and Physiochemical Properties
- 2.5.1 Project Hope server (http://www.cmbi.ru.nl/hope/) was used to search protein 3D structures by collecting structural information from a series of sources, including calculations on the 3D coordinates of the protein, sequence annotations from the UniProt database, and predictions by DAS services [35]. Furthermore, it described the reaction and physiochemical properties of these candidates. Protein sequence and mutant variants were submitted to project hope server in order to analyze the structural and conformational variations that have resulted from single amino acid substitution.2.5.2 Mutpred was used to predict structural and functional changes as a consequence of amino acid substitution. (http://mutpred.mutdb.org/). These changes were expressed as probabilities of gain or loss of structure and function. In addition, it predicts molecular cause of disease. The MutPred output contains a general score (g), i.e., the probability that the AAS is deleterious/ disease-associated and top five property scores (p), where p is the P-value that certain structural and functional properties are impacted. A missense mutation with a MutPred (g) score > 0.5 could be considered as “harmful,” while a (g) score > 0.75 should be considered a high confidence “harmful” prediction [36]. The input was submitted in the form of protein FASTA sequence along with amino acid substitution.
2.6. Prediction of Genetic and Protein Interactions
- Protein-protein interactions are important to assess all functional interactions among cell proteins. 2.6.1 STRING (Search Tool for the Retrieval of Interacting Genes/Proteins; https://string-db.org/) [37]. The STRING database gives a protein- protein interaction either it is direct or indirect associations. The input option we use is the protein name and the organism.2.6.2 KEGG (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/) [38] is a knowledge base for systematic analysis of gene functions in terms of the networks of genes and molecules, including metabolic pathways, regulatory pathways, and molecular complexes for biological systems.
3. Results
3.1. SNP Dataset from dbSNP
- As per dbSNP database, the human CALR gene investigated in this work contained a total of 682SNPs: 53 SNPs in 3′ UTR region, 25 SNPs in 5′ UTR region, 343 SNPs in intron region, 103 SNPs in coding synonymous regions and 154 non-synonymous SNPs (nsSNPs) that comprises of 150 missense mutations, 3 frameshift mutations and one nonsense mutation. The missense nsSNPs were selected for the investigation.
3.2. Prediction of Functional Mutations
- A total number of 150 nsSNPs CALR gene were submitted as batch to the SIFT program. According to our SIFT analysis predicted, 26 SNPs were predicted to be deleterious, 48 SNPs were predicted to be tolerated and 76 nsSNPs were not found. Deleterious SNPs were submitted to Polyphen-2, PROVEAN, SNAP and Condel.Polyphen-2 results analysis predicted that 9 nsSNPs were probably damaging, whereas 2 nsSNPs were predicted to be ‘possibly damaging’ and the remaining one nsSNPs was categorized as benign (Table 1). Our PROVEAN analysis predicted that 10 nsSNPs were deleterious and 2 nsSNPs were neutral. The SNAP2 Analyzer predicted that 9 nsSNPs were affected the function of protein and 3 nsSNPs were neutral (Table 1). Out of 26 nsSNPs predicted to be deleterious by SIFT, 12 nsSNPs were classified by the other computational tools used and 14 nsSNPs were unclassified.
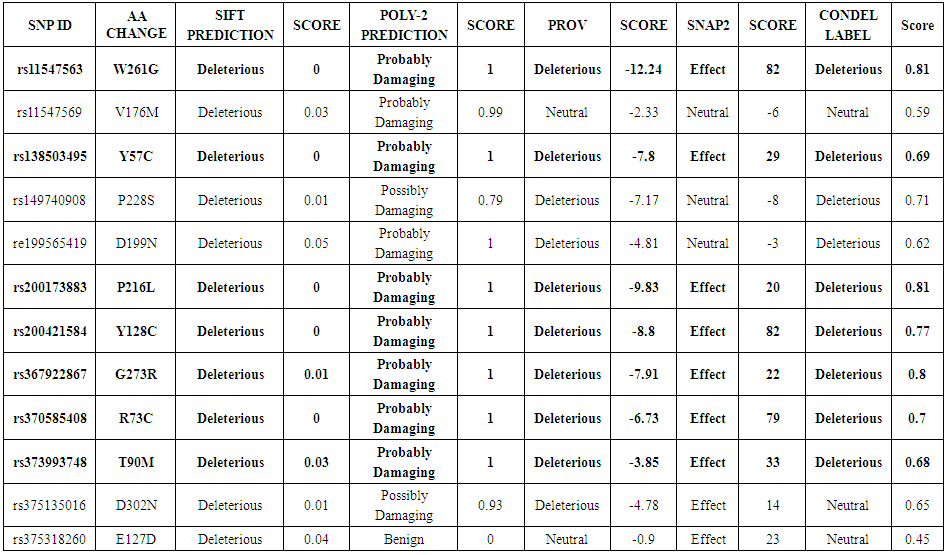 | Table 1. List of nsSNP analysis by SIFT, PolyPhen-2, PROVEAN, SNAP2, CONDEL |
3.3. Prediction of Disease Associated SNPs
- Pmut, nsSNPAnalyzer and PhD-SNP were used to validate the results obtained from five previous tools. Out of 7 nsSNPs that predicted to be deleterious with SIFT, Polyphen, SNAP2, PROVEAN and CONDEL; Pmut predicted all 7 nsSNP to be associated with disease, nsSNPAnalyzer predicted 6 nsSNP while nsSNPAnalyzer predicted 5 nsSNP to be associated with disease (Table 2). Finally, out of 26 nsSNP, 4 nsSNPs namely rs11547563 (W261G), rs200173883 (P216L), rs200421584 (Y128C) and rs370585408 (R73C) were found to be highly significant deleterious by (SIFT, Polyphen, PROVEAN, SNAP2, CONDEL, Pmut, nsSNPAnalyzer and PhD-SNP) servers.
|
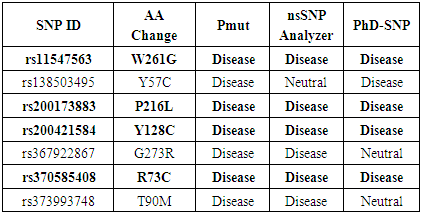
3.4. Prediction of nsSNPs Impact on the Protein Stability
- All SNPs predicted to be associated with disease were submitted to I- mutant server to predict the impact of this SNPs on the protein Stability. The results were as follows: 3 SNPs (W261G, Y128C, R73C) were predicted to decrease effective stability of the CRT protein and one SNP (P216L) was predicted to increase the stability of CRT protein (Table 3).
|
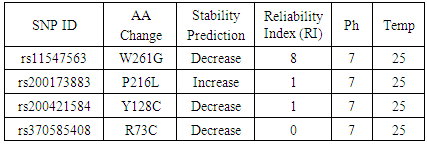
3.5. Analysing the Effect of nsSNPs on 3D Structure of the Proteins and Physiochemical Properties
- 3.5.1 3D structural modelling by Project Hope serverrs11547563 (W261G) The residue is located in the P-domain of the protein; this domain is important for binding of other molecules and in contact with residues in N-terminal region domain that is also important for binding. Mutation of this residue might disturb the interaction between these two domains and as such affect the function of the protein. The mutation introduces a glycine at this position. Glycine are very flexible and can disturb the required rigidity of the protein at this position. The mutant residue is smaller and less hydrophobic than the wild-type residue. The wild-type residue forms a hydrogen bond with Valine at position 50. The size difference between wild-type and mutant residue makes that the new residue that is not in the correct position to make the same hydrogen bond as the original wild-type residue did. Moreover, this difference in hydrophobicity will affect hydrogen bond formation. (Figure 2).
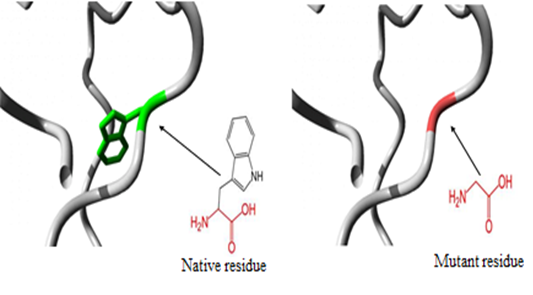 | Figure 2. Close-up of the mutation. The protein is colored grey, the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. SNP ID: rs28989182, protein position 261changed from Tryptophan to Glycine |
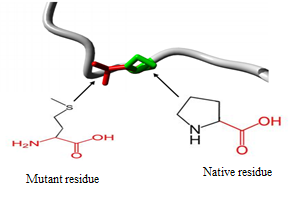 | Figure 3. Close-up of the mutation. The protein is coloured grey, the side chains of both the wild-type and the mutant residue are shown and coloured green and red respectively. SNP ID: rs200173883, protein position 216 changed from Proline to Leucine |
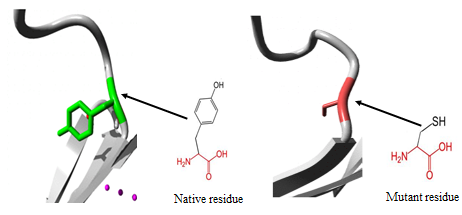 | Figure 4. Close-up of the mutation. The protein is colored grey, the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. SNP ID: rs200421584, protein position 128 changed from Tyrosine to Cysteine |
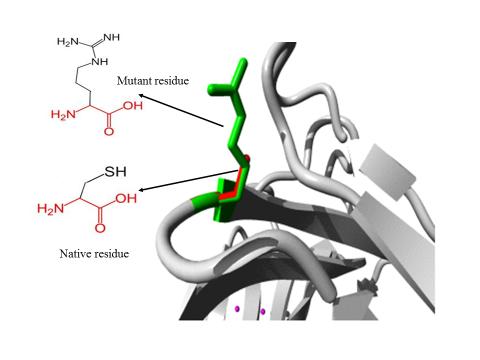 | Figure 5. Close-up of the mutation. The protein is colored grey, the side chains of both the wild-type and the mutant residue are shown and colored green and red respectively. SNP ID: rs370585408, protein position 73 changed from Arginine to Cysteine |
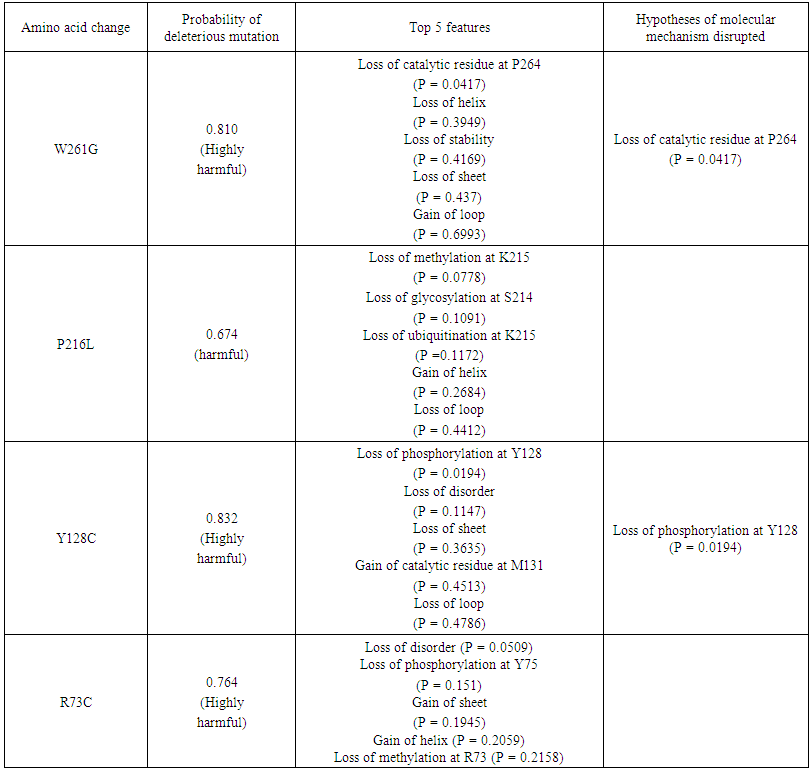 | Table 4. Prediction of functional effects of nsSNPs using MutPred |
3.6. Protein-Protein Interactions Analysis
- STRING interaction analysis revealed that CALR gene involved in many molecular and biological process and has high-confidence interactions with Heat shock protein 90kDa beta member 1 (HSP90B1), Protein disulfide isomerase family A member 3 (PDIA3), Heat shock 70kDa protein 5 (HSPA5), Prolyl 4-hydroxylase, beta polypeptide (P4HB), Glucosidase, alpha; neutral AB (GANAB), Protein kinase C substrate 80K-H (PRKCSH), TAP binding protein (TAPBP), Apolipoprotein B (APOB), Activating transcription factor 6 (ATF6) and Beta-2-microglobulin (B2M) (Figure 6).
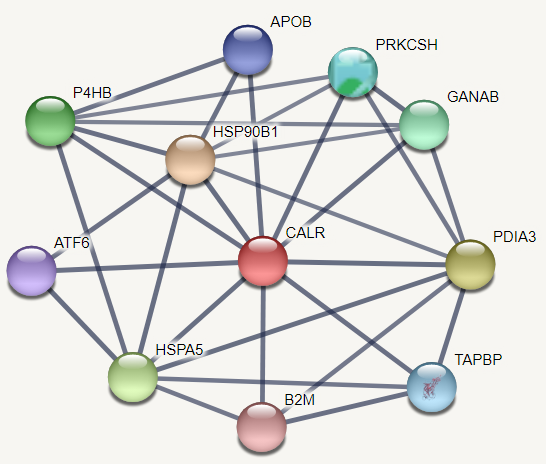 | Figure 6. Functional interaction between CALR and its related genes |
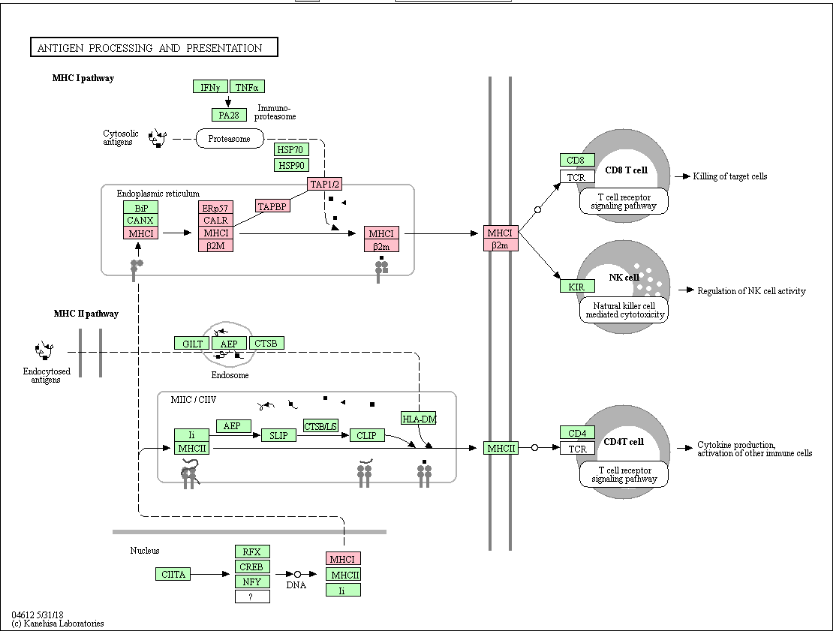 | Figure 7. Antigen processing and presentation network by MHC class I (KEGG ID: hsa04612) |
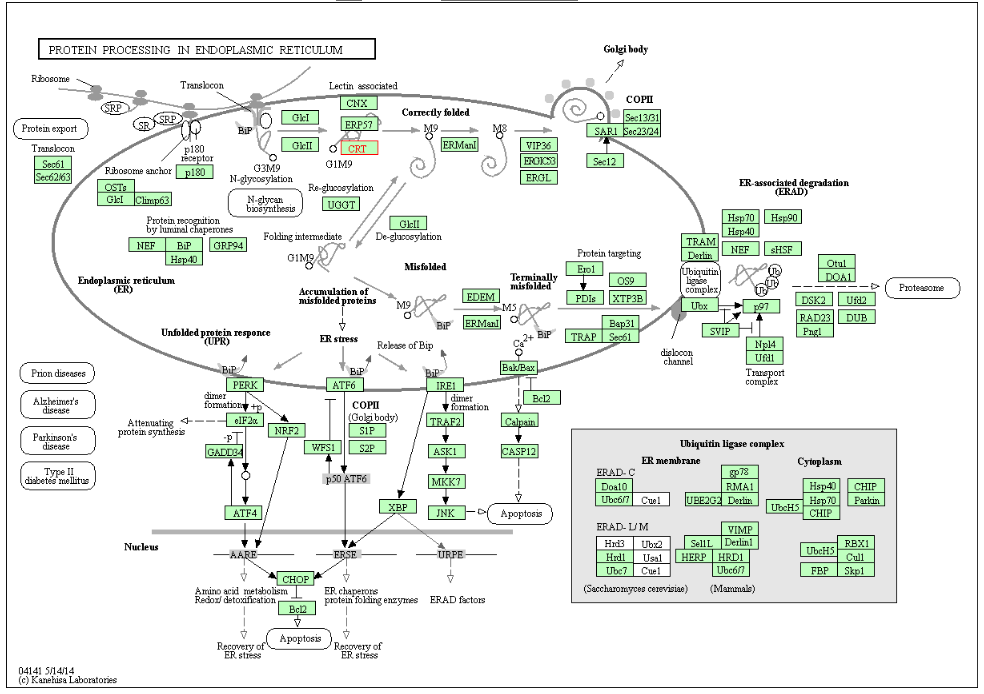 | Figure 8. Protein processing in endoplasmic reticulum (KEGG ID: hsa04141) |
4. Discussion
- The present study undertakes a systematic approach to identify functionally important nsSNPs in human CALR gene in an attempt to understand how do these mutations affect the protein structure and function and hence promote a disease. To sort out tolerant from intolerant nsSNPs eleven different prediction algorithm were used; SIFT, Polyphen, PROVEAN, SNAP2, CONDEL, Pmut, SNP Analyzer and PhD-SNP. By comparing the scores of all 8 methods, 4 nsSNPs with positions W261G, P216L, Y128C and R73C, W261G were found to be highly damaging nsSNPs and those were selected for further analysis by other in silico tools.Decreased stability of protein changes was observed in all deleterious SNPs except P216L. Most of the diseases causing and cancer- related mutations are found to destabilize the corresponding protein. Decreasing in protein stability was reported in VWF A2 domain causing von Willebrand disease type 2 [39] and it was found that N214D mutation could cause a defect in tumor suppressor protein ING4 (N214D) associated with lung carcinoma [40]. Meanwhile, increased protein stability was reported to be accompanied by an elevated protein levels and protein dysregulation. Such cases have included mutations in the CLIC2 protein (H101Q) that is associated with a mental disorder [41] and in DNMT1 protein associated with breast cancer [42].The possible molecular mechanism by which a deleterious variation associated with disease state was predicted by Mutpred. The majority of predicted effects were alteration in phosphorylation, loss of methylation, loss of catalytic residue and altered disorderness in protein structure. There is an evidence in the literature that loss of phosphorylation is to be associated with esophageal cancer [43] and loss of methylation sites are associated with hepatocellular carcinoma [44] and colorectal tumors [45].Furthermore, mutation 3D was used to investigate the location of nsSNPs in CRT domains which resulted in variations in amino acid properties (charge, size, flexibility and hydrophobicity value). Due to these polymorphisms, new version of mutant protein might be produced which then may affect functions and structure of the original protein.From STRING protein-protein interaction analysis, CALR was predicted with the strong interactions with HSP90B1, PDIA3, HSPA5, P4HB, GANAB, PRKCSH, TAPBP, APOB, ATF6 and B2M. The STRING interaction result was further validated by using KEGG pathways for CALR. Interestingly, same set of proteins were involved in protein processing in endoplasmic reticulum and antigen processing and presentation network by MHC class I molecules. Our literature search demonstrated that HSP90B1, HSPA5, PRKCSH, TAPBP, APOB, ATF6, B2M are involved in pathways of prostate cancer (46), Prion diseases (47), Polycystic liver disease (48), bare lymphocyte syndrome (BLS) type1 (49), familial hypercholesterolaemia (50), Achromatopsia (51) and familial hypercatabolic hypoproteinemia (52) respectively.Therefore, any changes in the CRT protein function would have an impact on many pathways, involved in diseases.
5. Conclusions
- Nowadays, in silico analysis has got major concern to screen diseases related nsSNP at molecular level. In this study a systematic and extensive in silico analysis of functional nsSNPs in the CALR gene has been performed to investigate the effect of nsSNPs on structure and function of CRT protein. Four nsSNPs with positions P216L, R73C, W261G and Y128C were predicted to be the most damaging mutations for CRT protein altering physiochemical properties of the protein; size, charge, hydrophobicity and stability leading to loss or disturbance of the protein internal and external interactions and eventually loss of the protein's function as well as disease association. Further, 16 SNPs at the 3UTR were predicted to introduce a change in the micro RNA binding site at the 3UTR which might result in deregulation of the gene function. This is computational approach, thus, population genetics based studies with clinical evidences are necessary to accompaniment the findings of this study.
ACKNOWLEDGEMENTS
- The authors acknowledge Dr. Ahmed Abdelbagi Hammad for language editing and proofreading of the manuscript.