-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2017; 7(1): 9-24
doi:10.5923/j.bioinformatics.20170701.02

Bioinformatics Analysis of Single Nucleotide Polymorphism in Multiple Drug Resistance Protein 1 (ABCB1) and Multi-Drug Resistance-Associated Protein 1 (ABCC1) in Bos taurus
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLNuha A. Mahmoud1, Elham I. Mohammed2, Siham O. Elshafei1, Ahmed H. Elsadig3, Rihab A. Omer4, Sofia B. Mohamed4
1Department of Biochemistry and Molecular Biology, Faculty of Medicine, National University, Sudan
2Faculty of Medical Laboratory Science, National University, Khartoum, Sudan
3Faculty of Medicine, University of Khartoum, Khartoum, Sudan
4National University Research Institute, Khartoum, Sudan
Correspondence to: Sofia B. Mohamed, National University Research Institute, Khartoum, Sudan.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The ABCB1 and ABCC1 genes are a transmembrane active efflux pump for a variety of drugs. They are a putative mechanism of multidrug resistance in a range of diseases. It is postulated that ABCB1 and ABCC1 single nucleotide polymorphism (SNP) contribute to variability in protein functions, and therefore in drug resistance, prognosis, toxicity and disease. However, there are not much attention has been given to SNP in ABCB1 and ABCC1 genes in Bos taurus. In this current study, a comprehensive analysis of the functional and structural impacts of all known SNPs of the genes, was performed using publicly available computational prediction tools. Two different databases were used to retrieve SNP data sets followed by five different in silico algorithms (SIFT, PROVEAN, PANTHER and GO& SNAP2) to screen nsSNPs in ABCB1 and ABCC1 genes. The comparative analysis predicted that 9 and 2 functional SNPs showed damaging effects in ABCB1 and ABCC1 subsequently. The ExPASy-PROSIT program revealed that the deleterious mutations (G516A and V560G) are contained in the ABC Transporter 2, (F707C) in ABC_TM1F domains, (K424E and C422W) in ATP binding cassette in ABCB1. And the (R719W) lay in the ABC_ Transporter2 domain for ABCC1. As well as identifying the putative functional SNPs that may possibly undergo post-translation; it was founded that mutation at K424E can also lead to an alteration in the ubiquitylation site within the ABCB1. The mutation (V1244G) of ABCC1 is also present at a sum interacting motif (SIM). Additionally, MUpro and I. Mutant showed a decrease in stability for high-risk nsSNPs in ABCB1 and ABCC1, suggesting that variants could destabilize the amino acid interactions directly or indirectly. This effect will cause to some extent, functional deviations of proteins. Furthermore, the 3D structure of both proteins using a CPH server and Chimera were also modeled. Finally, the 27 SNPs in 3’ un-translated region was analyzed using an mrSNP tool in an ABCB1 gene the results showed that there were no SNPs lay in 3’UTR. Throughout the review of this study; damaging effect nsSNPs in ABCB1 and ABCC1 genes were detected, and they were altered the structure and function of these genes. They are also may have a relation with ABCB1 and ABCC1 functions in Bos taurus. Recent advances understanding the role and the mechanism of ABCB1 and ABCC1 polymorphisms Tauruss taurus by Next Generation Sequencing are presented.
Keywords: ABCB1 ABCC1, Non-synonymous single nucleotide polymorphism (nsSNP), 3’UTR SNPs, Exons, Bioinformatics analysis, Multiple drug resistant, and Bos taurus
Cite this paper: Nuha A. Mahmoud, Elham I. Mohammed, Siham O. Elshafei, Ahmed H. Elsadig, Rihab A. Omer, Sofia B. Mohamed, Bioinformatics Analysis of Single Nucleotide Polymorphism in Multiple Drug Resistance Protein 1 (ABCB1) and Multi-Drug Resistance-Associated Protein 1 (ABCC1) in Bos taurus, American Journal of Bioinformatics Research, Vol. 7 No. 1, 2017, pp. 9-24. doi: 10.5923/j.bioinformatics.20170701.02.
Article Outline
1. Introduction
- ABC (ATP-binding cassette) proteins from one of the largest protein families; members of these families are found in all the living organisms starting from microbes to humans. Limited numbers of ABC transporters play a role in drug and metabolite transportation; ABCB1 (ATP-binding cassette sub-family B member 1), ABCC1 (ATP-binding cassette sub-family C member 1), ABCC2 (ATP-binding cassette sub-family C member 2), ABCC4 (ATP-binding cassette sub-family C member 4) and ABCG2 (ATP-binding cassette sub-family G member 2), although these numbers are likely to grow. These transporters are localized in the plasma membranes of the intestine, liver, kidney, blood-brain barrier and other vital biological barriers, thus explaining their effect on the disposition of drugs. Glycoprotein P (MDR1/ABCB1) and multidrug resistance protein 1 (MRP1/ABCC1) are ATP-binding cassette (ABC) multidrug-efflux pumps that play an important role in normal physiology in protecting tissues from toxic xenobiotics and endogenous substrates such metabolites [1]. Expression of these efflux transporters in the gastrointestinal tract and brain capillary endothelial cells limits oral absorption and central nervous system uptake of many drugs [2]. Moreover, ABCB1 and ABCC1 transporters involvement in drug transport through cell membranes is likely contributing to a variety of drug disposition and response [1]. ABCB1 and ABCC1 play a major role in the anthelmintic drug resistant macrocyclic lactone endectocides (MLS), such as Ivermectin. The increasing knowledge of the ABCB1 transports involved in anthelmintic resistance will play an important role in allowing the identification of markers to monitor the spread of resistance and to evaluate new tools and management practices aimed at delaying its spread [3]. These proteins play essential roles in the delivery of, for example, vitamins, fatty acids, sterols, porphyrins, thyroid hormones, carnitine, and amino acids to the breast-fed offspring [4]. However, some of these transporters feature broad substrate specificities and have been demonstrated to mediate active transport of drugs and toxic chemicals into milk [5]. ABC transporters in veterinary medicine are evident to be widely used as licensed drugs such as; digoxin, verapamil, loperamide, cimetidine, ivermectin, macrolides and fl macrolides. Chemotherapeutic agents in cancer therapy are increasingly being applied to related animal species due to the increased inclination of pet owners for advanced therapies [6]. Single nucleotide polymorphisms (SNPs) in genes that are encoding the ABC drug-efflux pumps, may play a role in responses to drug therapy and disease susceptibility [7, 8]. The contribution of ABCB1 to multidrug resistance is well established and this attributed to its overall structure and function, which has been studied extensively [1] since its discovery in 1976 [9]. The effect of the various genotypes and haplotypes on the expression and function of these proteins is not yet clear, also their true impact remains controversial [7]. Additionally, ABCB1 variants have been implicated in the etiology of several human diseases associated with resistance to pharmacotherapy [2]. Two common polymorphisms in the ABCB1 gene (C3435T and G2677T/A/C) have been associated with differences in gene expression and protein activity, and with variability in disposition of many therapeutic drugs as well [10, 11]. The C3435T (rs1045642) is a synonymous polymorphism, (Ile1145) located in the exon 26 that was found to be associated with expression levels of ABCB1 [10]. Polymorphisms of ABCB1 in other animal species have been found as a result severe phenotypes. For example, a 4-bp deletion in the canine ABCB1 exon leads to a frameshift mutation. The collies that carry both mutant alleles become hypersensitive to ivermectin, a deworming agent, because they lack functional ABCB1 transporters [12]. Another example is found in a subpopulation of CF-1 mice, comprising 25% of the animals, which display much higher drug sensitivity to ivermectins [13, 14]. Indeed, new SNPs have recently been identified, and the list is expanding. The associations of P-gp expression with drug resistance have also been shown in canine lymphoma. The transduction of the canine ABCB1 gene has been reported to induce resistance to several chemotherapeutic agents in the canine cell line [15] Even though most of the SNPs have low frequencies, they may still affect the function of ABCB1, either alone or in conjunction with other SNPs. ABCC1 is expressed in many tissues, and function as an efflux transporter for glutathione-, glucuronate- and sulfate conjugates as well as unconjugated substrates. It can also deliberate resistance to a broad range of chemotherapeutic agents and transport a variety of toxicants [11]. Sequence variations within the ABCC1 gene might account for differences in drug response in different individuals. More than 85 polymorphisms have been identified in ABCC1 [16]. The ABCC1 G2012T (rs45511401) is a non-synonymous SNP, (Gly671Val) located in the exon 16 at the nucleotide binding domain of the protein [17]. G2012T SNP which occurs at low frequency. By which only one or two of the four populations examined were predicted to be functionally deleterious and hence are likely to be under negative selection [15, 18]. This SNP may be useful for the studies associating ABCC1 variants with rare events including adverse drug reactions. In veterinary medicine, it was shown that ABCC1 was responsible for vinblastine and cisplatin resistance in canine mammary tumor cell lines [19], and the expression of the ABCC1 gene was generally detected in all samples of the 103 primary canine mammary tumors [20]. Thus, polymorphism is likely an important feature of ABCB1 and ABCC1 in disease susceptibility, drug response and treatment outcomes [21]. Therefore, studying haplotypes of ABCB1 and ABCC1 could increase our understanding of the structure and function of these genes. However, nsSNP and miRNA SNPs analysis for ABCB1 and ABCC1 in Bos tarus have not been estimated computationally until now. In this work, different, publicly available, bioinformatics tools and databases for a comprehensive analysis of nsSNPs and mirSNPs in ABCB1 and ABCC1 were employed. Since the majority of disease mutations affect protein stability, we also proposed modeled protein structures of the mutant proteins and compared them with the native protein in order to evaluate stability changes.
2. Material and Method
2.1. Retrieval of SNPs
- Polymorphism data for the ABCB1 and ABCC1 genes were retrieved from the UniProt database (http: //www. uniprot.org) and the NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/SNP/).The SNP database contains SNPs 3’/5′ UTR, 3′/5′ splice sites, intron, missense, nonsense, stop gained and frame shift. In this study, the Bos taurus SNPs within the coding (non-synonymous) and 3'′ UTR had been selected and submitted to the bioinformatics tools for further investigation.
2.2. Non-synonymous SNP Analysis
- The functional effects of nsSNPs on the ABCB1 and ABCC1 genes were predicted using the following in silico algorithms: SIFT, PROVEAN, SNAP2, SNPs & GO and PANTHER, in order to test its deleterious effect. The nsSNPs which were analyzed by the SIFT tool was taken for more analysis of different algorithms.
2.2.1. Deleterious nsSNP Found by SIFT Program
- SIFT is a sequence homology-based tool that predicts the variation of the protein function caused by the change in the amino acid sequence (http://sift.jcvi.org/). The hypothesis states that the positions which are important for the function of protein should be conserved in the protein family, whereas insignificant positions should not be conserved. The SIFT scores were classified as damaging (0.00–0.05), or tolerant (0.201–1.00) [22]. SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence (http://sift.jcvi.org). It is a multi-steps procedure that, for a given protein sequence searches for similar sequences were done, and then closely related sequences that may share a similar function were chosen. The multiple alignment of these chosen sequences was obtained, and the normalized probabilities for all possible substitutions at each position of the alignment were calculated [23].
2.2.2. Predication of Functional Impact of nsSNP by PROVEAN
- PROVEAN (Protein Variation Effect Analyzer) predicts the functional impact for all classes of protein sequence variations not only for single amino acid substitutions, but also insertions, deletions, and multiple substitutions on the alignment-based score. The score measures the change in sequence similarity of a query sequence to a protein sequence homology between with and without an amino acid variation of the query sequence. If the PROVEAN score is ≤-2.5, the protein variant is predicted to have a "deleterious" effect, whereas if the PROVEAN score is >-2.5, the variant is predicted to have a "neutral" effect [24].
2.2.3. Effected and Neutral nsSNP Predicted by SNAP2
- SNAP2 is a bioinformatics tool classifies the genetic variation based on the neutral network, which predicts the changes due to the nsSNPs on the secondary structure. It also compares the solvent accessibility of the native protein and mutated one, in order to distinguish the min to effect (+100, strongly predicted) or the neutral effect (-100, strongly predicted). The FASTA sequence of the native and ABCB1 and ABCC1 proteins were provided as an input, the SNAP2 provided a heat map with possible substitution at each position of these proteins, where, when the score is greater than >50 in the dark red indicates strong signals for pathogenicity. (https://rostlab.org/services/snap/) [25].
2.2.4. Validation and Functional Characterization Predications by SNPs & GO and PANTHER
- SNPs & GO is also a support vector machine (SVM) based on the method to accurately predict the mutation related to the disease from protein sequence. The input is the FASTA sequence of the whole protein, while the output is based on the difference between the neutral and disease related variations of the protein sequence. The RI (reliability index) with a value greater than 5 depicts indicates that the disease-related effect is caused by the mutation of the functional part of the parent protein. The PANTHER algorithms were also used in the display of the output [26].
2.3. Protein Stability Analysis
- Protein stability of the mutant ABCB1 and ABCC1 were predicted using the following in silico algorithms: I-mutant 2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0.html) and MUpro (http://www.ics.uci.edu/~baldig/mutation. Html).
2.3.1. Investigation of Mutation Protein Stability by I-Mutant 2.0 and MUpro Prediction
- I-Mutant 2.0 is an SVM based tool i.e., Support vector machine based tool which leads to an automatic protein stability change prediction which causes a single point mutation. The initiations were done either by using the protein structure or more precisely; were taken from the protein sequence. The output is a free energy change value (ΔΔG), Positive ΔΔG value infers that the protein being mutated is of higher stability than the negative ΔΔG value and vice versa. MUpro is also a support vector machine-based tool for the prediction of protein stability changes upon non-synonymous SNPs. The value of the energy change was predicted by measuring the confidence score between -1 and 1. A score less than <0 means the variation decreases the protein stability; conversely, a score >0 means the variance increases the protein stability [27].
2.4. Prediction of nsSNPs Positions in Different Protein Domains Using PROSITE Database
- PROSITE, is a protein domain database for functional characterization and annotation. It consists of documentation entries describing protein domains, families, and functional sites, as well as associated patterns and profiles to identify them. It is complemented by ProRule, which is a collection of rules based on profiles, and patterns, which increases the discriminatory power of these profiles and patterns by providing additional information about functionally and/or structurally critical amino acids. PROSITE is largely used for the annotation of domain features of UniProtKB/ Swiss-Prot entries [28].
2.5. Prediction of Post-Translational Modification Sites
- UbPred (www.ubpred.org) and BDM-PUB (bdmpub.biocuckoo.org) programs are used to predict the ubiquitylation sites [29]. In UbPred, lysine residues with a score of 0.62 were considered ubiquitylated. For BDM-PUB, the balanced cut-off option was selected. SUMOplot (http://www.abgent.com/sumoplot) and SUMOsp 2.0 (http://sumosp.biocuckoo.org/) programs were used to predict the sumoylation sites [30]. For SUMOplot, only high probability motifs with a score .0.5 were considered sumoylated. Medium level threshold with a 2.64 cut-off value was selected for SUMOsp 2.0 analysis. In addition, Putative phosphorylation sites were also predicted using the GPS 2.1 (http://gps.biocuckoo. org/) and NetPhos 2.0 (http://www.cbs.dtu.dk/services/ NetPhos/) [31]. For the GPS 2.1 analysis, high-level threshold with cutoff values ranging from 0.776 to 11 were selected. In NetPhos 2.0, serine, threonine, and tyrosine residues with a score of .0.5 were considered phosphorylated. Sumo-interacting motifs (SIM) were identified manually and they were compared to experimentally verified SIMs in the scientific literature [32].
2.6. Presentation and Visualization of Proteins
- Biological sequence diagrams are fundamentals for visualizing various functional elements in protein or nucleotide sequences that enable a summarization and presentation of existing information as well as means of intuitive new discoveries.
2.6.1. Illustrator of Biological Sequences (IBS)
- IBS is the software used for representing the organization of protein or nucleotide sequences in a convenient, efficient and a precise manner. Multiple options are provided in IBS such as; biological sequences which can be manipulated, recolored or rescaled in a user-defined mode. Also, the final representational artwork can be directly exported into a publication-quality figure. (http://ibs.biocuckoo.org/online.php) [33].
2.7. Molecular Effects of High-Risk nsSNPs on Protein Structure
- Three-dimensional (3D) structural analyses were done for ABCB1 and ABCC1 proteins (wild type) with each of the high-risk nsSNP located within proteins. Homology modeling was done by Web-based server CPH.
2.7.1. CPH Models
- A protein homology modeling prediction server was used to predict the 3D structure of proteins using an unknown 3D structure model. In CPH models the template recognition is based on a profile to profile alignment guided by secondary structure and exposure predictions. Protein sequence requirements were submitted to the CPH server to get the model as a PDB file for the structure that could not be predicted by the automated Project Hope server. The resultant PDB files were opened using Chimera program which was used to visualize the PDB structure. (http://www.cbs.dtu.dk/services/CPHmodels/) [34].
2.7.2. Modeling Amino Acid Substitution; H-Bonding and Clash
- UCSF Chimera is a highly extensible program for interactive visualization and analysis of molecular structures and related data. The Chimera (version 1.8) software was used to scan the 3D (three-dimensional) structure of a specific protein, and hence the original amino acid was modified with the mutated one to see the produced impact with the outcome then being a graphic model depicting the mutation. Chimera (version 1.8) currently available within the Chimera package and available from the Chimera web site (http://www.cgl.ucs f.edu/chimera/) [35].
2.8. Detection of SNP Effects on microRNA Binding by Using the mrSNP Software
- The mrSNP Software analyzes the functional impact of genetic polymorphisms (SNPs) within the micro RNAs binding sites. The mrSNP is a highly adaptable and performing tool for predicting what the effect of a 3’UTRSNP on miRNA-binding. This tool has an advantage over existing algorithms as it can assess the effect of the novel SNPs on miRNA-binding without requiring significant hands-on time. (http://compbio.uthsc.edu/miRSNP/) [36].
3. Results
3.1. Variant Calling
- Polymorphism data for the ABCB1 and ABCC1 genes were retrieved from the NCBI dbSNP database and the UniProt database. According to this database, the ABCB1 gene was found to have188 missense and 27 SNPs in its 3' UTR. The dbSNP of ABCC1 gene also found to have 245 missenses with no SNPs found in the 3' UTR. To determine whether a given mutation affected ABCB1 and ABCC1 function, tsubjectse mutation was subjected only to a variety of in silico SNP prediction algorithms. In addition, the SNPs in 3'UTR found in the ABCB1gene were analyzed by bioinformatics tools.
3.1.1. Deleterious and Effect of nsSNPs by SIFT, PROVEAN and SNAP2
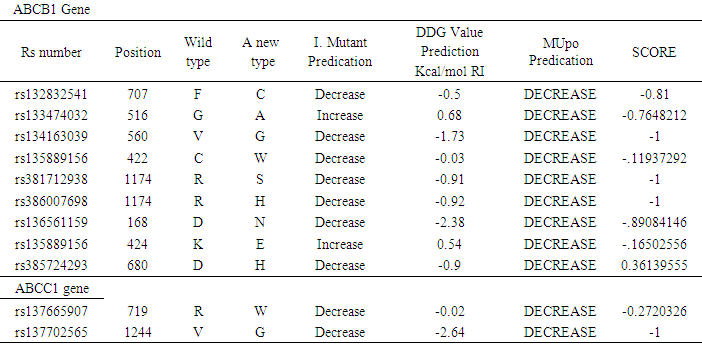
- In ABCB1gene, SIFT analyzed 27 nsSNPs out of 188 in the database, and in ABCC1 7 nsSNPs out of 245 was analyzed. These nsSNPs were taken for more analysis by different tools. The result showed that out of 27 nsSNPs in MDR1, 7 (F707C, V560G, K424E, C422W, R1174S, D680H, R1174H) were identified to be deleterious and effected by SIFT, PROVEAN and SNAP2, 3nsSNPs (G516A, D168N and L536V) were identified deleteriously by SIFT and PROVEAN. Two nsSNP (F723L and A718) were predicted deleterious by PROVAEN. One nsSNP (I1276V) was predicted deleterious by SIFT. And two nsSNPs (H1245R and R1186H) were identified effected by SNAP2. In ABCC1, out of 7 missense SNPs, 2 (R719W and V1244G) were identified deleterious and effect by SIFT, PROVEAN and SNAP2. The nsSNPs with triple tools predication and the score for ABCB1 and ABCC1 genes are shown in (Table 1).
|
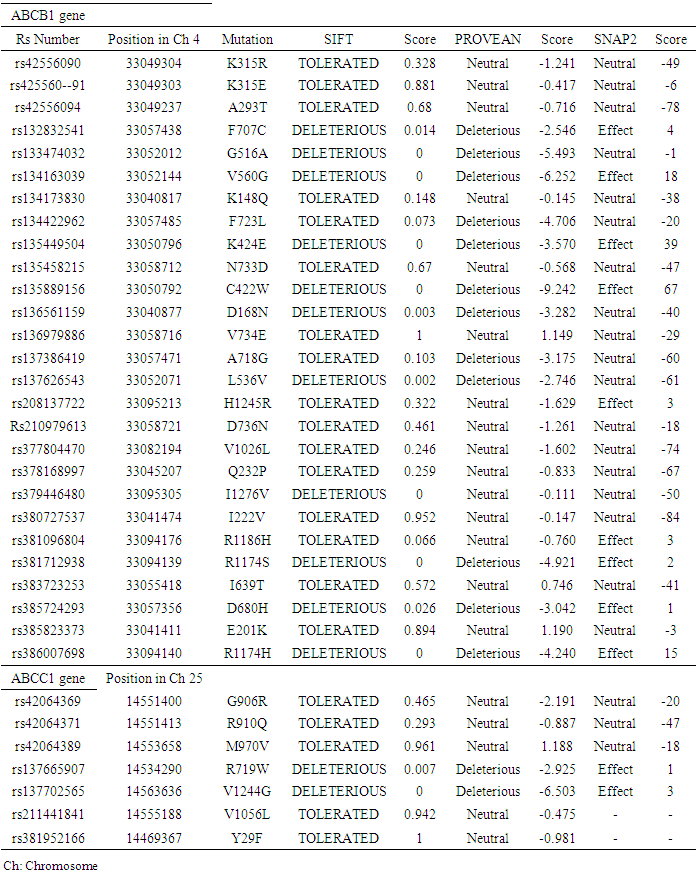
3.1.2. Functional Prediction of nsSNPs Using PANTHER and SNPs&GO
- Further confirmation of the effects of the 27 nsSNPs in ABCB1 and the 7 nsSNPs in ABCC1 were analyzed using PANTHER and SNPs&GO tools. In ABCB1 9 nsSNPs were predicted diseases caused by PANTHER and SNPs&GO (F707C, G516A, V560G, F723L, K424E, C422W, V1026L, R1174S, R1174H), and 6 nsSNPs (D168N, A718G, L536V, Q232P, D680H and E201K) were classified diseases caused by PANTHER. In ABCC1 two nsSNPS were predicted diseases caused by PANTHER and SNPs&GO (R719W and V1244G). A summary of the results can be found in (Table 2).
|
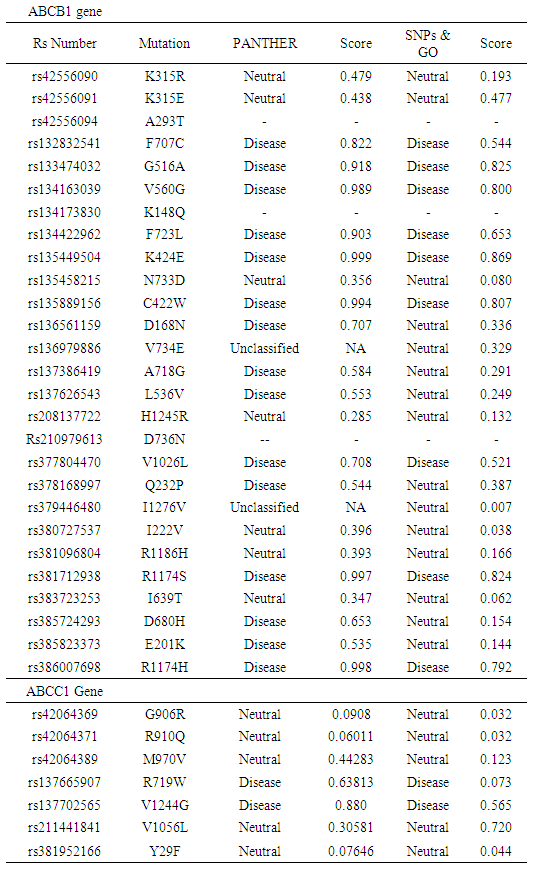
3.2. Mutant Amino Acids Affect the Domain Structures of ABCB1 and ABCC11 by Using the PROSITE Database
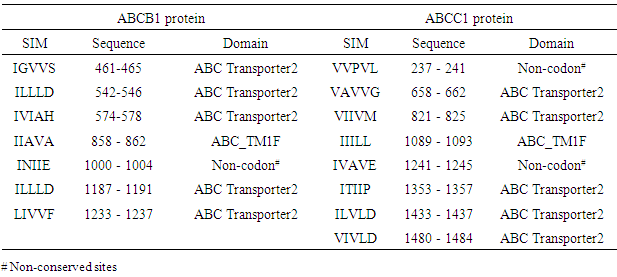
- Further investigations of the potential effects of the 9 high-risk nsSNPs in ABCBI and the 2 high-risk nsSNPs in ABCC1 were made using the PROSITE database for predicting the domain region. The IBS online program was then used to distribute the nsSNPs within the domain. The results show, ABC transporter integral membrane type-1 (ABC_TM1F) and ABC_ transporter 2), as well as one NP-BIND (binding site) ATP binding cassette. The results were shown in (Table 3).
|
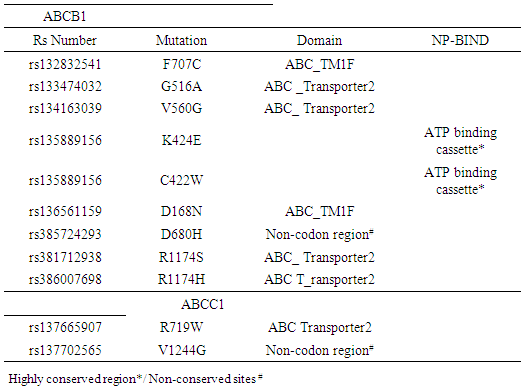
3.3. Prediction of Post-Translational Modification Sites in ABCB1 and ABCC1
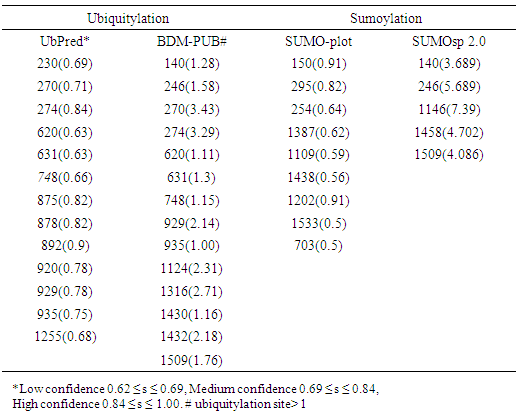
- To analyze the residues in ABCB1 and ABCC1 that may undergo ubiquitylation or sumoylation, the UbPred BDM-PUB, SUMO-plot, and SUMOsp 2.0 programs were used. UbPred predicted that 7 lysine residues in ABCB1 undergo ubiquitylation. In contrast 10 lysine residues that undergo ubiquitylation were predicted by BDM-PUB. Residues (K1018) that undergo ubiquitylation were predicted by both UbPred and BDM-PUB. By using BDM-PUB; one putative PTM was coincided in location with nsSNPs in the ABCB1 gene (K424E). SUMOplot predicted that 10 lysine residues in ABCB1 were sumoylated whereas SUMOsp 2.0 predicted that 4 lysine residues were sumoylated (Table 4). UbPred predicted that 13 lysine residues in ABCC1 were ubiquitylated. In contrast, BDM-PUB predicted that 14 lysine residues were ubiquitylated. Both UbPred and BDM-PUB predicted that residues (K270, K274, K620, K631, K929 and K935) were ubiquitylated. SUMOplot predicted that 9 lysine residues in ABCC1 were sumoylated, whereas SUMOsp 2.0 predicted that 5 lysine residues sumoylated (Table 4). In addition, to putative sumoylation sites, 7 potential sumo-interacting motifs (SIM) in ABCB1were identified and 8 in ABCC1. One nsSNP (V1244G) in ABCC1 protein was found in SIM (IVAVE) (Table 6).
|
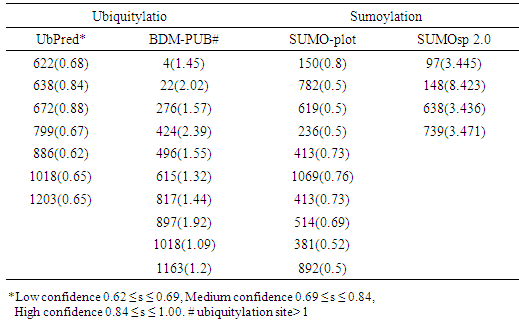
|
|
3.4. Prediction of Phosphorylation Site in ABCB1 and ABCC1 Proteins
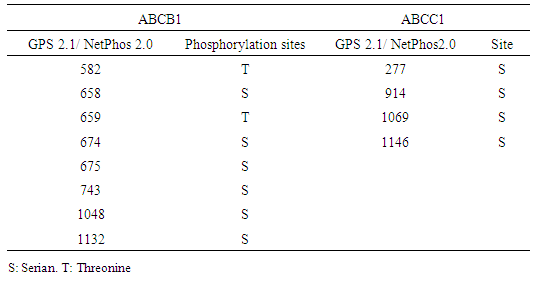
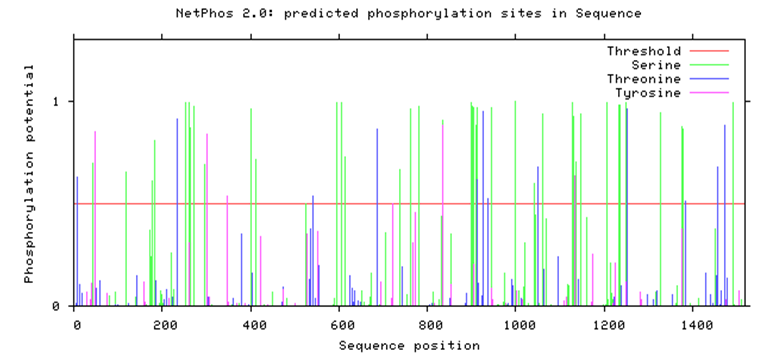
- The GPS 2.1 and NetPhos 2.0 servers were used to predict Serine, Threonine, and tyrosine phosphorylation sites in the proteins. 6 Serine (S) -specific phosphorylation sites and 3 Threonine (T) -specific sites in the ABCB1 protein were predicted by the GPS 2.1 server conversely, 35 Serine-specific phosphorylation sites, 19 Threonine-specific sites, and 9 Tyrosine-specific sites were predicted by NetPhos 2.0. 8 Serine-specific phosphorylation sites and 2 Threonine-specific sites in the ABCC1 protein were predicted by the use of the GPS 2.1 server. Conversely, 45 serine-specific phosphorylation sites, 12 Threonine-specific sites, and 55 Tyrosine-specific sites were predicted by the Netplus 2.0. In ABCB1, Six serine residues and 2 Threonine residues were predicted to be phosphorylated by both GPS 2.1 and NetPhos 2.0 servers. In ABCC1, 4 Serine residues were predicted to be phosphorylated by both GPS 2.1 and NetPhos 2.0 servers (Table 7). None nsSNPs were found on Phosphorylation site in ABCB1 and ABCC1 proteins.
|
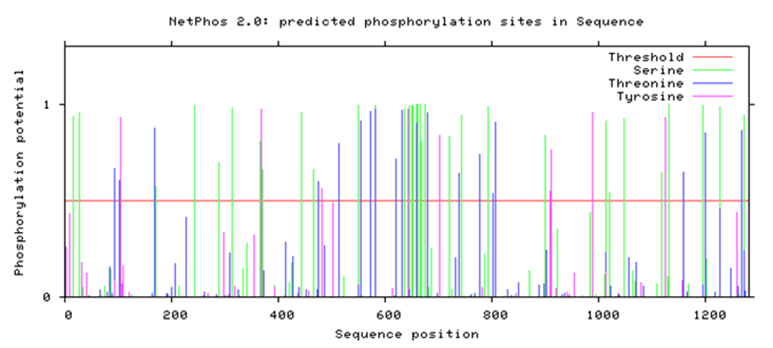 | Figure 1. Predicted phosphorylation sites in ABCB1 by Netphos 2.0 |
 | Figure 2. Predicted phosphorylation sites in ABCC1 by Netphos 2.0 |
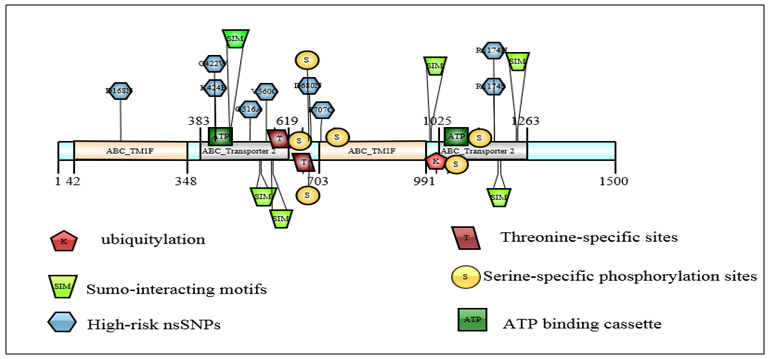 | Figure 3. Schematic depiction of the approximate ABCB1 protein locations of the top predicted PTM sites (ubiquitylation and sumoylation,), the 9 high-risk nsSNPs and the 6 sumo-interacting motifs (SIMs). Several domains of known functional importance are marked on the protein (top image), including the ABC_TM1F and ABC_Transporter 2 domains, as well as the NP_BIND ATP binding cassette (required for bind nucleotide phosphates) |
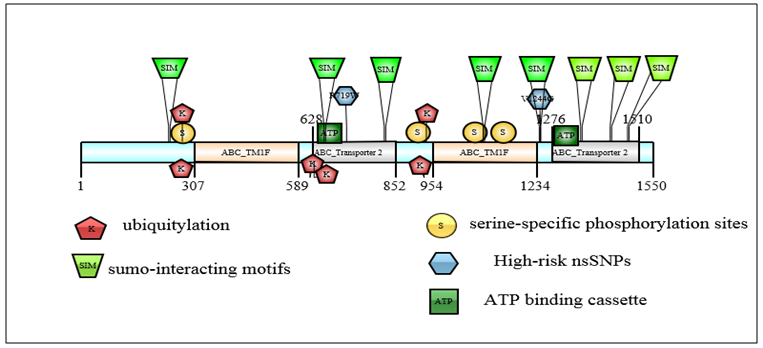 | Figure 4. Schematic depiction of the approximate ABCC1 protein locations of the top predicted PTM sites (ubiquitylation, sumoylation, and phosphorylation), the 2 high-risk nsSNPs and the 8 sumo-interacting motifs (SIMs). One high-risk nsSNP site (V1244) was predicted to undergo SIM in the wild type of protein. Several domains of known functional importance were marked on the protein (top image), including the ABC_TM1F and the ABC_Transporter 2 domains, as well as the NP_BIND ATP binding cassette (required for bind nucleotide phosphates) |
3.5. Prediction of Change in Protein Stability due to nsSNPs
- To add another layer of confirmation, the effects of these nsSNPs were analyzed deleterious by at least 4 algorithms using I. Mutant and MUpro. In ABCB1 gene, all nsSNPs (F707C, G516A, V560G, K424E, C422W, D168N, R1174S, D680H and R1174H) showed a decrease resulting in the stability of the protein by MUpro and one nsSNPs showed an increase result by I. Mutant. In the ABCC1 gene, all nsSNPs showed a resulting decreasthe stabilityility of the protein by I. Mutant. All nsSNPs with the DDG value and score for I. Mutant and MUpo of ABCB1 and ABCC1 were shown in (Table 8).
|
3.6. 3D Structure Modeling of ABCB1 and ABCC1
- The 3-dimensional structure expectation CPH models 3.2 server was utilized. Visualization and characterization of the proteins model were done by Chimera (version 1.8) program. The result is shown in figure 5 (A and B).
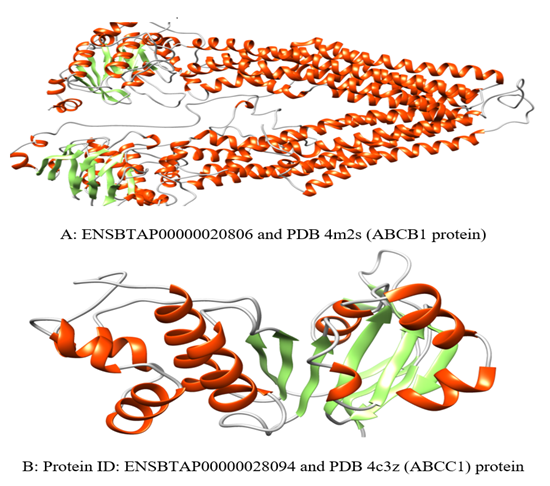 | Figure 5. A and B show the protein tertiary structure backbone and their protein secondary structures (alpha helix, Beta sheet and random coil) used CPH models 3.2 server and Chimera software. The ID number and PDB below pictures show related to protein sequence records in Uniprot database and Protein data bank respectively |
3.7. Comparative Modeling of High-risk Non-synonymous SNPs
- Theoretical structural models were generated for each nsSNP using the Chimera program, which constructs 3D models for proteins based on homologues of known structure. The H-bonding interactions and clashes of wild type and mutant of nsSNPs were calculated using the Chimera program. The native and mutant protein structure for the 9 nsSNPs of ABCB1 and only one ABC (SNP(R719W), due to the structure in the Protein data bank, is desiwithed by a CPH server that has a sequence less than 1244 Aacids acid. The nsSNP of ABCB1 and ABCC1 are shown in figure (6-12).
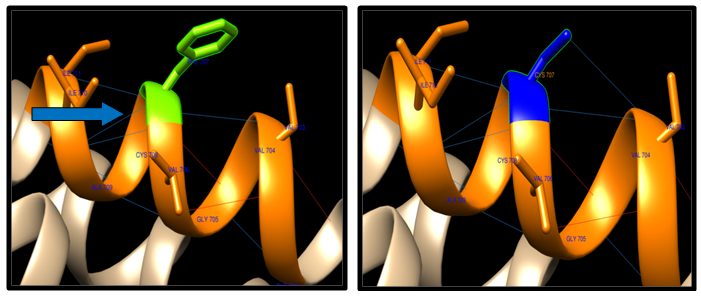 | Figure 6. F707C SNP structure: 10 H-bonding interaction in wild type and 11 in mutant. Wild type green color and mutant blue color |
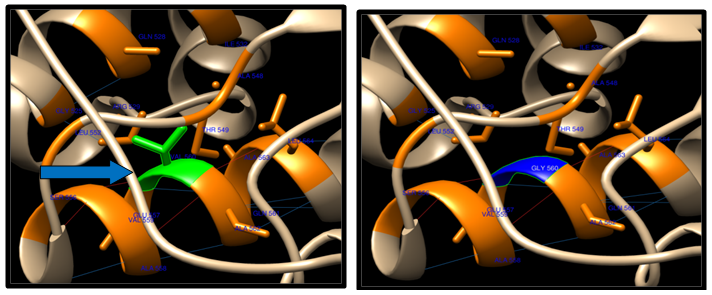 | Figure 7. V560G SNP structure 15 H-bonding interaction in wild type and 13 in mutant. Wild type green color and mutant blue color |
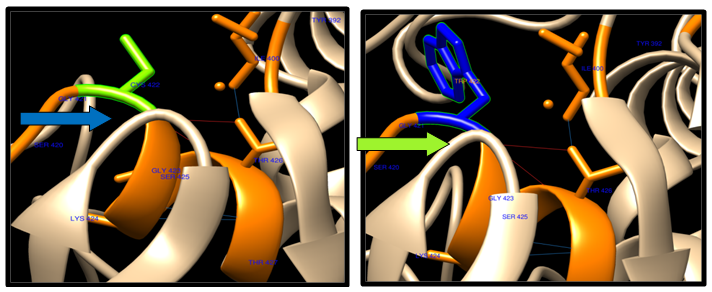 | Figure 8. C422W SNP structure 9 H-bonding interaction in wild type and mutant. Wild type green color and mutant blue color |
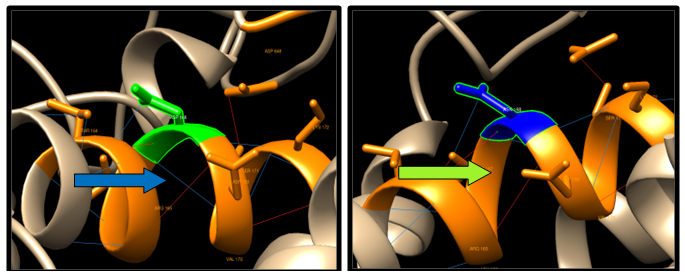
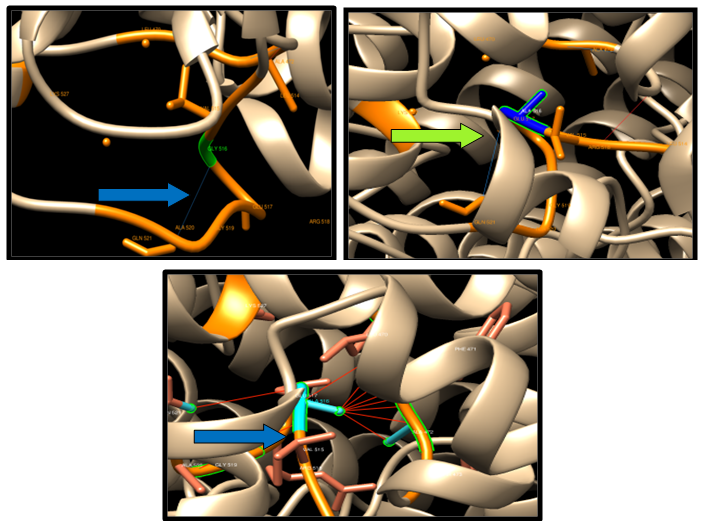 | Figure 9. G516A SNP structure 5 H-bonding interaction in wild type and in mutant. Wild type green color and mutant blue color. 10 clashes were detected between the mutant and the Amino Acid |
 | Figure 10. D168N SNP structure 16 H-bonding interaction in wild type and in 14 mutants. Wild type green color and mutant blue color |
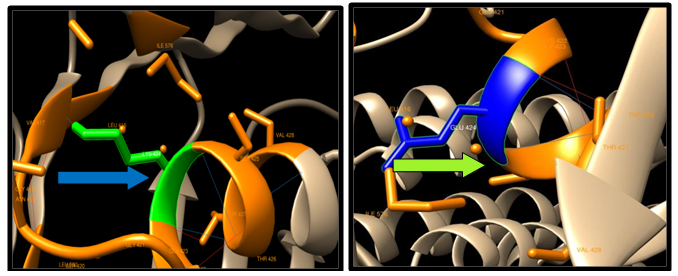 | Figure 11. K424E SNP structure 12 H-bonding interaction in wild type and 9 in mutant. Wild type green color and mutant blue color |
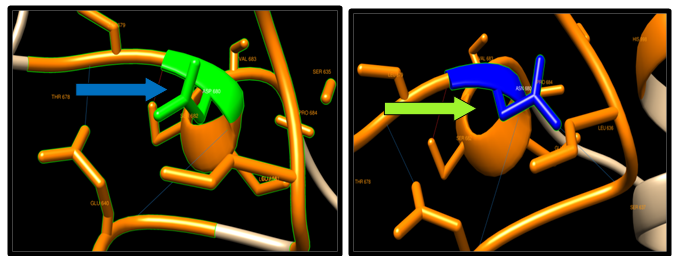 | Figure 12. D680N SNP structure 3 H-bonding interaction in wild type and in 4 mutants. Wild type green color and mutant blue color |
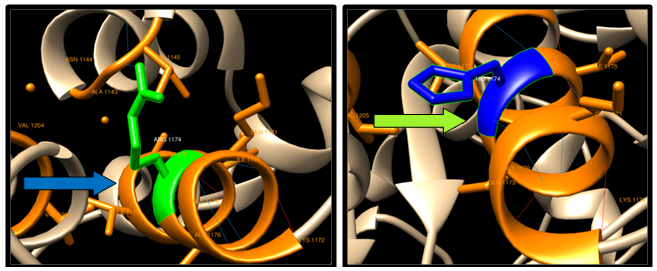 | Figure 13. R1174H SNP structure: 12 H-bonding interaction in wild type and 10 in mutant. Wild type green color and mutant blue color |
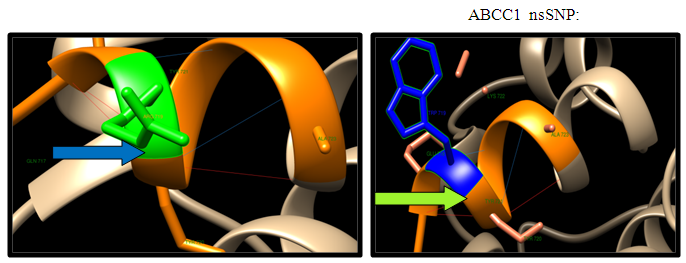 | Figure 14. R719W SNP structure 5 H-bonding interaction in wild type and 5 in mutant. Wild type green color and mutant blue color |
3.8. Scanning of Functional SNP in Untranslated Regions (UTR) Predicted by mrSNP
- Single variants within 3'UTR were selected from dbSNP and NCBI and submitted to the mrSNP server, to check whether these variants could disrupt or create new miRNA binding sites or have no impact at all. A total of 27 SNPs in ABCB1 were founded and analyzed by mrSNP with the result showing that no SNP lay in the 3'UTR (Table 9).
|
4. Discussion
- In this study, we have examined the ABCB1 and ABCC1 proteins in Bos taurus in order to analyze and identify the deleterious and functional nsSNPs using in silico methods. ABCB1 and ABCC1 are one of the associated proteins in which the two shares closely related domain structures to both the ABC_MT1F and ABC transporter 2 domains and ATP binding site. The functional effects of nsSNPs were predicted using the following in silico algorithms; SIFT, PROVEAN, SNAP2, PANTHER and SNPs&GO. Each algorithm uses different parameters to evaluate the nsSNPs with more positive results more likely to be truly deleterious. The nsSNPs predicted to be deleterious by at least four in silico algorithms categorized as high-risk nsSNPs which were selected for further analysis. Nine nsSNP were found in ABCB1 protein deleterious by at least four in silico algorithms (F707C, V516G, C422W, G516A, R1174S, R1174H, D168N, K424E and D680H), and 2 nsSNPs in ABCC1 (R719 and V1244G). The amino acids that are involved in important biological processes, such as those located in enzymatic sites or required for protein interactions, tend to be more conserved than other residues. As such, nsSNPs that are located at highly conserved amino acid positions tend to be more deleterious than nsSNPs that are located in non-conserved sites [36]. In ABCB1 mutation studies have confirmed that changes in crucial amino acid residues in the trans-membrane domains, ATP-binding domains, Walker-A motifs, Walker-B motifs, or the signature motif affect ABCB1 function [37, 38]. Mutations may also affect folding pathways or protein conformation [39]. For example, non-synonymous amino-acid substitutions in trans-membrane5 (TM5) or trans-membrane (TM6) affect the ability of UIC2, a conformational-sensitive antibody, to bind to ABCB1 [39]. In ABCC1, some SNPs (e.g., T1684C and G4002A) also vary according to ethnicity [40], and may be linked to functional changes. In the present study, in ABCB1 we found 4 deleterious nsSNPs (G516A, V560G, R1174H and R1174S) located in the ABC Transporter 2 domain, 2 deleterious nsSNPs (F707C and D168N) located in the ABC_TM1F domain, 2 deleterious nsSNPs (K424 and C422W) located in the ATP binding cassette. Additionally, in ABCC1 one deleterious nsSNP (R719W) was found located in ABC Transporter 2. These nsSNPs may have a functional significance because they reside on the surface of regions of the protein that are potentially important such as the ATP binding, or reside in domains region. Post-translational modification sites (PTMs) are involved in many biological processes, including a number of canonical innate immune pathways, and they are essential for the regulation of the protein structure and function [41]. To investigate how nsSNPs may influence (PTMs) of ABCB1 and ABCC1, we used a variety of in silico prediction tools to identify putative PTM sites in the proteins. The results showed one nsSNP (K424E) in ABCB1 predicted to undergo ubiquitylation site by a BDM-PUB server. Sumo-interacting motifs SIMs are short hydrophobic motifs that interact non-covalently with other sumoylated proteins. In putative sumoylation sites, we identified one nsSNP (V1244G) potential sumo-interacting motifs (SIM) in ABCC1 by SUMOsp 2.0. Also, these SNPs are the most likely to affect ABCB1 and ABCC1 activities. Accurate prediction of protein stability changes resulting from single amino acid mutations is important for understanding protein structures and designing new proteins. Hence, I-Mutant and MUpro were used to predict whether deleterious nsSNPs altered the stability of the ABCB1 and ABCC1 proteins. Out of 9 ABCB17, nsSNPs decreases the stability of protein, and both nsSNPs in ABCC1 were predicted to be less stable than the wild type protein. The high-risk nsSNPs (K424E) in ABCB1 located at putative PTM sites, also decreased the ABCB1 protein stability by MUpro. The high-risk nsSNPs (V1244G) in ABCC1 located on the SIM site decreased protein stability by MUpro and I. Mutant. A number of studies have shown that decreased protein stability leads to increased protein misfolding, aggregation, and degradation. To this end, the 9 nsSNPs in ABCB1 and one nsSNPs in ABCC1 had the H- bond and clash shown between the wild type and mutant using the visualized Chimera program. In ABCB1 7 nsSNPs out of 9 (F707C,516G, R1174S, R1174H, D168N, K424E and D680H) have a differing number of H-bonding between wild and mutant residues indicating that mutation will affect the protein stability, as well as clashes detected in (V516G) mutant structure indicating the change in the environment of the protein which may affect the structure and function. To examine whether nsSNPs altered the 3D structure in ABCB1 and ABCC1, we individually substituted each nsSNP into the wild type ABCB1 and ABCC1 sequences and then the sequences were submitted to Chimera for structural analysis. From the results; we found all nsSNPs altered the 3D structure of the proteins, so this may affect the protein functions. SNPs in the UTR sites are involved in the regulation of gene expression in many ways, such as RNA transcript splicing site or transcription factor binding site alteration [42]. In human, miR-451, miR-27a, miR-21, miR- 130a, miR-let-7, miR-137, miR-200c, miR-122, miR-138 and miR-10a/b was suggested to regulate ABCB1 gene expression indirectly by targeting other mRNAs that code the proteins associated with the activation of ABCB1 gene expression [43, 44]. Additionally, lymphoblastic leukemia (ALL) cell lines, miR-455-3p was shown to be related to P-gp expression level acute lymphoblastic leukemia (ALL) cell lines, miR-455-3p was shown to be related to P-gp expression level [45]. However, there has been no study on the regulations of ABCB1 gene or P-gp expression by miRNAs in veterinary medicine. Hence, the UTRs were also analyzed for their functional SNPs. The result explained all SNPs didn’t lay in the 3'UTR. Based on these observations, we concluded that these SNPs should be considered important candidates in causing anti- resistant drug, prognosis, toxicity and disease in Bos taurus. A closer insight in localization and functional of SNPs in ABCB1 and ABCC1in cattle will allow the prediction of clinical relevance of substrate interaction and the development of new pharmaceuticals tailors, made to reach pre-selected tissues, such as the mammary gland, for the treatment of common and economically important disease in cattle. Moreover, it is important to identify SNPs in ABCB1and ABCC1 and examine their effect on protein function. Also experimentally study to support this insilico analysis needs.
5. Conclusions
- Our results demonstrate that multiple nsSNPs in the ABCB1 and ABCC1 proteins may be deleterious to proteins structure and/or function by different bioinformatics tools. Most of these high-risk nsSNPs are located at highly conserved amino acid sites in an ABC Transporter 2 and ABC_ TM1F domains, as well as the ATP binding cassette. In addition to these findings, we also identified several ABCB1 and ABCC1 sites that may undergo post-translational modification, including sites that coincide with the location of high-risk nsSNPs. In this study, we show that K424E of the top high-risk nsSNPs disrupt the Putative ubiquitylation site of ABCC1 in ATP binding cassette domain. Based on our results, we concluded that these SNPs should be considered important candidates in causing diseases related to ABCB1 and ABCC1 proteins. This study is the first systematic and extensive in silico analysis of functional SNPs in the ABCB1 and ABCC1 in Bos taurus.
ACKNOWLEDGEMENTS
- This work was carried out with the support of the National University Research Institute.