-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2016; 6(2): 56-97
doi:10.5923/j.bioinformatics.20160602.03

Computational Analysis of Deleterious Single Nucleotide Polymorphisms (SNPs) in Human MutS Homolog6 (MSH6) Gene
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLNahla E. Abdelraheem1, Marwa Mohamed Osman2, Osama Muhieldin Elgemaabi3, Afra Abdelhamid Fadl Alla2, Mosab Mohamed Ismail2, Soada Ahmed Osman2, Aisha Ismail Ibrahim2, Nihad Elsadig Babekir4, Salwa Osman Mekki5, Mohamed A. Hassan2, 6
1Department of Histopathology and Cytology, Faculty of Medical Laboratory Sciences, National University, Khartoum, Sudan
2Department of Biotechnology, Africa City of Technology, Khartoum, Sudan
3Department of General Surgery, Omdurman Military Hospital, Omdurman, Sudan
4National Center for Neurological Sciences (NCNS), Khartoum, Sudan
5Deparment of Pathology, Faculty of Medicine, Alneelin University, Khartoum, Sudan
6Division of Molecular Genetics, Institute of Human Genetics, University of Tubingen, Germany
Correspondence to: Nahla E. Abdelraheem, Department of Histopathology and Cytology, Faculty of Medical Laboratory Sciences, National University, Khartoum, Sudan.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Background: Point mutations in MSH6 gene had been related to group of cancers called lynch syndrome which accounts for 3% to 5% of all colorectal cancers. Despite the excessively studied MSH6 mutations, the mechanism by which these mutations promote carcinogenesis remains controversial. Methods: MSH6 was investigated in dbSNP/NCBI in December 2015, 3666 SNPs were found in human; 388 were coding synonymous, 937 non synonymous, 201 in frame shift, 63 in 3' un-translated region and 347 in 5' un-translated region. Non synonymous and 3'UTR SNPs were selected for insilico analysis; SIFT, Polyphen2, Imutant3.0, MUpro, PhD-SNP, SNPs & GO, MutPred, ELASPIC, Mutation 3D, UCSF Chimera 1.8, PolymiRTs, and GENEMAIA softwares and servers were used to investigate the effect of SNPs on MSH6 protein's structure and function. Results: 21 SNPs were found to be highly damaging for the protein by SIFT and Polyphen, and were further analyzed by I-Mutant, MUpro, PHD-SNP, SNPs & GO, ELASPIS, Mutation 3D and Chimera. 2 SNPs were predicted by PolymiRTs to induce disruption or creation of miR binding sites; rs200412142 contained 2 disrupting and 1 creating functional classes in 3 miRSite, while rs184571821 SNP contained 3 creating functional classes in 3 miRSites. GENEMANIA revealed five genes similar in their expression level with MSH6 and seven genes share the same protein domain with it. Conclusions: 14 nsSNPs (R1217K, H1248D, E1214A, T1219I, K1140R, A1303T, M1137T, A1303G, R915K, H946D, T917I, A1001T, L899F, A1001G) were located at the interface of the MSH6 protein interfering with its relation with MSH2ISO2, MSH3, MSH2 and E9PHA6. Interactions of MSH6 with these proteins are critical for its MMR function and any structural alterations that interfere or harm these networks interactions would probably increase susceptibility to tumors formation and progression. 2 SNPs at the 3UTR; rs200412142 and rs184571821 introduced a change in the micro RNA binding site at the 3UT which might result in deregulation of the gene function.
Keywords: Lynch syndrome, MSH6, SNPs, In Silico, SIFT, Polyphen2, MutPred, Genemania, Polymirts and UCSFChimera
Cite this paper: Nahla E. Abdelraheem, Marwa Mohamed Osman, Osama Muhieldin Elgemaabi, Afra Abdelhamid Fadl Alla, Mosab Mohamed Ismail, Soada Ahmed Osman, Aisha Ismail Ibrahim, Nihad Elsadig Babekir, Salwa Osman Mekki, Mohamed A. Hassan, Computational Analysis of Deleterious Single Nucleotide Polymorphisms (SNPs) in Human MutS Homolog6 (MSH6) Gene, American Journal of Bioinformatics Research, Vol. 6 No. 2, 2016, pp. 56-97. doi: 10.5923/j.bioinformatics.20160602.03.
Article Outline
1. Introduction
- Single nucleotide polymorphism (SNP) is the variation in a genetic sequence that affects only one of the basic building blocks—adenine (A), guanine (G), thymine (T), or cytosine (C)—in a segment of a DNA molecule and that occurs in more than 1% of a population. The DNA of humans may contain many SNPs, since these variations occur at a rate of one in every 100–300 nucleotides in the human genome. In fact, roughly 90 percent of the genetic variation that exists between humans is the result of SNPs. Although the majority of variations do not alter cellular function and thus have no effect, some SNPs have been discovered to contribute to the development of diseases such as cancer and to influence physiological responses to drugs. [1] An understanding of the relationship between these genetic variations and their phenotypic effects could therefore be a step toward exploring the causes of various disorders or diseases. SNPs can fall within the coding regions (coding SNPs) or noncoding regions of genes (non coding SNPs), or in the intergenic region between two genes [2, 3]. While the two others are quite natural in the human genome and phenotypically neutral [4], nonsynonymous coding SNPs (nsSNPs) are thought to have the principal impact on phenotype by changing the protein sequence. As they cause amino acid alteration in the corresponding protein product, it may exert deleterious effects on the structure, function, solubility, or stability of proteins [5]. Also the nsSNPs perturb gene regulation by modifying DNA and transcriptional binding factors [6] and the maintenance of the formational integrity of cells and tissues [7]. Thus, it is likely that nsSNPs play a major role in the functional diversity of coded proteins in human populations and often associated with human diseases. Indeed, studies have revealed that more than 50% of the mutations associated with inherited genetic disorders are resulted from nsSNPs [8]. Disease-causing mutations are frequently observed at either core or interface residues mediating protein interactions. Mutations at core residues frequently destabilize protein structure while mutations at interface residues can specifically affect the binding energies of protein-protein interactions. Missense mutations at protein–protein interaction sites, called interfaces, are important contributors to human disease. Interfaces are non-uniform surface areas characterized by two main regions, “core” and “rim”, which differ in terms of evolutionary conservation and physicochemical properties. Moreover, within interfaces, only a small subset of residues (“hot spots”) is crucial for the binding free energy of the protein–protein complex [9]. Protein-protein interactions are critical for nearly every process in the cell and deleterious mutations hindering these interactions can have severe consequences on the associated cellular function. A variety of efforts from personalized medicine to understand viral evolution require knowing how specific mutations effect the protein-protein interactions. Conversely, designing proteins with improved binding or altered specificity requires that the impact of mutations on the native interface be understood. Currently this information is not available experimentally on the proteome-wide scale necessary for these tasks. Considerable effort has been devoted towards developing methods to predict the impact of mutations on binding affinity. Most of these approaches rely on physics based methods that attempt to faithfully model on the atomic level the interactions determining protein-protein binding affinity. However, a major obstacle of such approaches is the need for the reconstruction of the full-atomic model for every mutant complex, which limits the accuracy of the approach (since the position of the side-chains is difficult to model) and reduces the computational speed and the range of applications (since rebuilding the full-atomic model is generally the most time-consuming step) [10]. MSH6 gene is located on chromosome 2 and consists of 13 exons. It encodes a member of the mismatch repair family (MMR). In E.coli; the MutS protein helps in the recognition of mismatched nucleotides prior to their repair. A highly conserved region of approximately 150 amino acids, called the walker-A adenine nucleotide binding motif exists in MutS homologs. The encoded protein heterodimerizes with MSH2 to form a mismatch recognition complex that functions as a bidirectional molecular switch that exchange ADP and ATP as DNA mismatches are bound and dissociated [11]. MSH6 gene mutations had been found to be involved in Lynch syndrome, leading to the production of an abnormally short, nonfunctional MSH6 protein or a partially active form of the protein. When the MSH6 protein is absent or nonfunctional, the number of mistakes that are left unrepaired during cell division increases substantially. The errors accumulate as the cells continue to divide, which may cause the cells to function abnormally, increasing the risk of tumors formation in the body [12]. In a MMR mechanism, the mismatch recognition function is fulfilled by one of the heterodimeric protein complexes, MSH2–MSH6 (MutSα) or MSH2–MSH3 (MutSβ), dependent on the type of mutation. The MutSα complex recognizes base–base mismatches and small insertion–deletion loops (IDL), whereas the MutSβ complex recognizes IDLs basically larger than one extra helical nucleotide [13-16]. The MSH6 and MSH3 proteins are shown to be functionally redundant, so that MutSα can partially compensate the function of MutSβ while MutSβ appears to only recognize insertions and deletions [17-19]. Because of this redundancy, mutations in MSH6 cause accumulation of base substitutions but less frequently frameshift mutations in microsatellite sequences [17, 20]. This explains the low microsatellite instability (MSI) in MSH6-deficient tumors. Lack of frameshift mutations, which can easily target repetitive sequences also in tumor suppressor genes and result in their inactivation during tumorigenesis, may further explain the late onset in many MSH6 mutation carriers. Lynch syndrome, also known as hereditary non polyposis colorectal cancer syndrome (HNPCC), accounts for 3% to 5% of all colorectal cancers and is an autosomal dominant inheritance, the susceptibility disorder is caused by germline mutations in mismatch repair (MMR) genes, 10% in MSH6 and PMS2 [2]. Carriers of MMR gene mutations are at high risk of early-onset colorectal and endometrial cancer. The Lynch syndrome includes tumors of the ovaries, small bowel, urothelium, biliary tract, and stomach [21, 22], and is generally suspected if there is familial aggregation of Lynch syndrome – associated cancers using criteria such as Amsterdam II or Bethesda [23, 24] or a tumor phenotype showing high DNA microsatellite instability [25]. MSH6 mutations are associated with many cancers; sporadic and hereditary colorectal cancer, endometrial cancer, prostate cancer, gastrointestinal cancers, childhood hematologic malignancies, glioblastoma, anaplastic oligodendroglioma and melanoma [26-34]. The number of mutations reported in the MSH6 gene is continually rising [35]. Families associated with MSH6 mutations unusually often display carcinomas of the endometrium [36-38]. The significance of MSH6 in endometrial carcinomas development is emphasized by the observation that lack of MSH6 protein characterizes endometrial but not colon carcinomas in HNPCC [39].Recently bioinformatics has became increasingly important in biology and bioinformatic tools aid in the processing and extraction of useful results from large amounts of raw data; textual mining of biological literature, analysis of gene and protein expression, simulation and modeling of DNA, RNA, and protein structures, comparison of genetic and genomic data and helps analyze and catalogue the biological pathways and networks that are an important part of systems biology. [40-43]. The International Collaborative Group on HNPCC (ICG-HNPCC) has over 30 potentially pathogenic MSH6 mutations in the database (http://www.nfdht.nl). A significant proportion (35%) of them results in a single amino acid substitution, which is difficult to interpret. The pathogenicity of HNPCC mutations is linked to malfunction of MMR. despite the association of MSH6 mutations with many types of cancers; the exact role of many reported mutations in tumorgenesis and cancer progression remain unknown. In this study we adopted an insilico approach to analyze human MSH6 reported mutations using different bioinformatics softwares to investigate the effect of single nucleotide polymorphisms on protein's structure and function and whether these variations can contribute in disease or not.
2. Material and Methods
- Different soft-wares; SIFT, polyphen-2, Imutant3.0, MUpro, PhD- SNP, SNPs & GO MutPred, ELASPIC, mutation 3D, GENEMANIA, PolymiRTs and chimera were used to investigate the effect of SNPs mutations on MSH6 protein structure and function. Prediction of deleterious effect of non synonymous SNPs was done by SIFT and Polyphen-2 soft-wares. Prediction of stability changes was investigated in I mutant-3 and MUpro. The association of nsSNPs with disease was done by PhD-SNP and SNPs & GO software. The structural changes in 3D structure including hydrogen bonding, clashes and contacts for each residue were analyzed using Chimera software. SNPs at the 3'UTR were analyzed to detect the effect on microRNA binding sites using PolymiRTs soft-ware. GENEMANIA was used to investigate MSH gene interactions. In this study we selected nsSNPs and those at the 3’UTR regions for analysis.
2.1. Investigation of MSH6 Gene's Interactions and Appearance in Networks in GENEMANIA Database GENEMANIA
- is an online database that helps in the prediction of gene function; it also finds other genes that are related to a set of input genes, using a very large set of functional association data that include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. It can also be used to find new members of a pathway or complex, find additional genes that may have been missed in screening or find new genes with a specific function, such as protein kinases. The question is defined by the set of genes in the input [44]. Available at: http://www.genemania.org/.
2.2. Prediction of Structural Impact of nsSNPs on Protein by SIFT software (v5.1)
- SIFT (Separating Intolerant from Tolerant): Is a sequence homology-based tool that sorts intolerant from tolerant amino acid substitutions and predicts whether an amino acid substitution in a protein will have a phenotypic effect. SIFT is based on the premise that protein evolution is correlated with protein function. Positions important for function should be conserved in an alignment of the protein family, whereas unimportant positions should appear diverse in an alignment. SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence. It is a multistep procedure that searches for similar sequences, then chooses closely related sequences that may share similar function to the query sequence, followed by obtaining the alignment of these chosen sequences, and finally calculates normalized probabilities for all possible substitutions from the alignment. [45] The input SNPs' rs-IDs were submitted to the server for analysis, positions with normalized probabilities less than 0.05 were predicted to be deleterious; those greater than or equal to 0.05 were predicted to be tolerated. Available at: http://sift.bii.a-star.edu.sg/.
2.3. Prediction of Deleterious nsSNPs by PolyPhen-2
- Polyphen (Polymorphism Phenotyping v2) is available as software and via a Web server. It predicts the possible impact of amino acid substitutions on the stability and function of human proteins using structural and comparative evolutionary considerations. It performs functional annotation of single-nucleotide polymorphisms (SNPs), maps coding SNPs to gene transcripts, extracts protein sequence annotations and structural attributes, and builds conservation profiles, then estimates the probability of the missense mutation being damaging based on a combination of all these properties. PolyPhen-2 features include a high-quality multiple protein sequence alignment pipeline and a prediction method employing machine-learning classification. The software also integrates the UCSC Genome Browser’s human genome annotations and MultiZ multiple alignments of vertebrate genomes with the human genome [46]. The input FASTA sequence of protein with the position of interest and the new residue were submitted to Polyphen to predict functional impact of mutations. Available at: http://genetics.bwh.harvard.edu/pph2/.
2.4. Analysis of nsSNPs' Impact on Protein Stability
- 2.4.1. I Mutant 3.0 Server: I-Mutant is a Support Vector Machine-based web server for the automatic prediction of protein stability changes upon single-site mutations starting from the protein structure or sequence. In both cases, it can predict the protein stability change corresponding to all possible mutations of a particular residue, or ask only for a specific mutation. In either case, I-Mutant3.0 can predict the direction of the free energy change and its value [47]. The input FASTA sequence of protein along with the residues changes were submitted to I mutant server for the analysis of DDG value (kcal/mol) and the RI value (reliabilityindex) was also computed. Available athttp://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/IMutant3.0.cgi.2.4.2. Mupro: Is a set of machine learning programs to predict how single-site amino acid mutation affects protein stability. It uses two machine learning methods: Support Vector Machines and Neural Networks. Both of them were trained on a large mutation dataset and show accuracy above 84% via 20 fold cross validation, which is better than other methods. One advantage of the method is that it do not require tertiary structures to predict protein stability changes [48, 49]. The value of the energy change is predicted, and a confidence score between -1 and 1 for measuring the confidence of the prediction is calculated. A score <0 means the variant decreases the protein stability; conversely, a score >0 means the variant increases the protein stability. Available at: http://www.ics.uci.edu/~baldig/mutation.html.
2.5. Prediction of Disease Associated Variations
- 2.5.1. PhD-SNP Software: PhD-SNP is Support Vector Machine based classifier that is optimized to predict if a given single point protein mutation can be classified as disease-related or as neutral polymorphism [50-53]. The input FASTA sequences of protein along with the residues change were submitted to PhD-SNP server for the analysis. Available at: http://snps.biofold.org/phd-snp/phd-snp.html.2.5.2. SNPs & GO: Is a support vector machine (SVM) based on the method to accurately predict the mutation related to disease from protein sequence. The input is the FASTA sequence of the whole protein, the output is based on the difference among the neutral and disease related variations of the protein sequence. The RI (reliability index) with value of greater than 5 depicts the disease related effect caused by mutation on the function of parent protein [54]. Available at: http://snps.biofold.org/snps-and-go/snps-and-go.html.
2.6. Prediction of Harmful Mutations by Mutpred
- The Mutpred server was employed to classify an amino acid substitution (AAS) as disease-associated or neutral. In addition, it predicts molecular cause of disease/deleterious AAS. Mutpred is based upon SIFT and a gain/loss of 14 different structural and functional properties. The output of Mutpred contains a general score (g), i.e., the probability that the amino acid substitution is deleterious/disease-associated, and top 5 property scores (p), where p is the P-value that certain structural and functional properties are impacted [55]. Available at: http://mutpred.mutdb.org/.
2.7. Prediction of the Stability Effects of Mutation in Domain Cores and Domain-Domain Interfaces
- ELASPIC is a novel ensemble machine learning approach that predicts the effects of mutations on protein folding and protein-protein interactions. Here we present the ELASPIC web server, which makes the ELASPIC pipeline available through a fast and intuitive interface. The web server can be used to evaluate the effect of mutations on any protein in the Uniprot database, and allows all predicted results, including modeled wild-type and mutated structures, to be managed and viewed online and downloaded if needed. It is backed by a database which contains improved structural domain definitions, and a list of curated domain-domain interactions for all known proteins, as well as homology models of domains and domain-domain interactions for the human proteome. Homology models for proteins of other organisms are calculated on the fly, and mutations are evaluated within minutes once the homology model is available [10]. Available at: http://elaspic.kimlab.org/many/.
2.8. Distribution of nsSNPs in MutS domains by Mutations 3D
- Mutation3D is a functional prediction and visualization tool for studying the spatial arrangement of amino acid substitutions on protein models and structures. It is intended to be used to identify clusters of amino acid substitutions arising from somatic cancer mutations across many patients in order to identify functional hotspots and fuel downstream hypotheses. It is also useful for clustering other kinds of mutational data, or simply as a tool to quickly assess relative locations of amino acids in proteins [56]. Available at: http://mutation3d.org/index.shtml.
2.9. Protein 3d Modeling and Detection of Hydrogen Bonding and Clashes by UCSF Chimera
- CPH models 3.2: is a protein homology modeling server. The template recognition is based on profile-profile alignment guided by secondary structure and exposure predictions [57]. Available at:http://www.cbs.dtu.dk/services/CPHmodels/.UCSF Chimera is a highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles [58]. Chimera (version 1.8) software was used to scan the 3D (three-dimensional) structure of specific protein, and hence modifies the original amino acid with the mutated one to see the impact that can be produced. The outcome is then a graphic model depicting the mutation. Chimera (version 1.8) is available within the Chimera package from the Chimera web site http://www.cgl.ucsf.edu/chimera/.
2.11. Prediction of the Impact of SNPs at the 3Un Translated Region (3UTR) by PolymiRTS Database
- PolymiRTS database: is designed specifically for the analysis of non-coding SNPs at 3'UTR. It identifies single-nucleotide polymorphisms (SNPs) that affect miRNA (micro RNA) targets in human and mouse [59]. We used this computational server to analyze 3'UTR SNPs in MSH6 gene that may alter miRNA binding on target sites resulting in diverse functional consequences. All SNPs located in that region were selected and submitted to PolymiRTS (v3.0), available at: http://compbio.uthsc.edu/miRSNP.
3. Results
3.1. Retrieving SNPs and Protein's Sequence from the Database
- A total of 7654 MSH6 SNPs were investigated in dbSNP/NCBI database (http://www.ncbi.nlm.nih.gov/snp) in December 2015; 3666 SNPs were in Homo sapiens; of which 388 were coding synonymous and 937 non synonymous SNPs, 201 were frame shift, 63 were in 3' un-translated region and 347 were in the 5' un-translated region. The FASTA formats of the protein (its isoforms and fragments) were obtained from Uniprot at Expassy database (http://expasy.org); the Uniprot accession numbers: (P52701, C9JH55 and C9J7Y7).
3.2. MSH6 Gene Interactions and Appearance in Networks
- Genemania revealed that MSH6 has many vital functions; a substantial role in DNA recombination, mismatch repair complex binding, negative regulation of DNA metabolic process, ATP metabolic process, DNA-dependent ATPase activity, reciprocal meiotic recombination, purine nucleoside monophosphate catabolic process, structure-specific DNA binding, purine, ATP catabolic process, nucleoside monophosphate catabolic process, coupled meiotic nuclear division, meiotic cell cycle, nucleoside monophosphate metabolic process, meiosis I, nuclear chromosome, double-stranded DNA binding, regulation of DNA metabolic process, DNA secondary structure binding, cellular process involved in reproduction, nuclear division, organelle fission, somatic diversification of immunoglobulins, somatic diversification of immune receptors, imunoglobulin production, production of molecular mediator of immune response, magnesium ion binding, isotype switching and response to radiation. The genes co-expressed with, share similar protein domain, or participate to achieve similar function are listed in table (7, 8 in the appendix) and Figure (1).
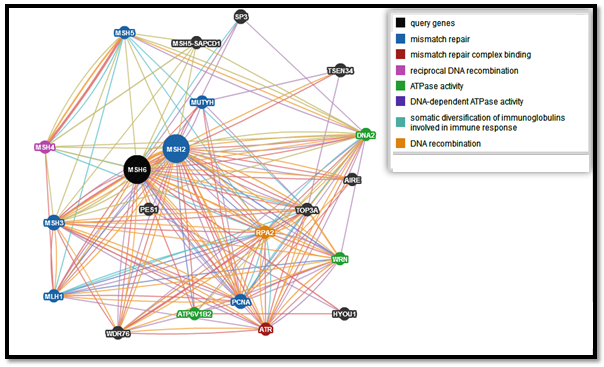 | Figure 1. Functional interactions between MSH6 and its related genes |
3.3. Prediction of Deleterious nsSNPs by SIFT and Polyphen
- Coding SNPs were analyzed using SIFT and Polyphen soft-wares. Batch nsSNPs (rs-IDs) were submitted to SIFT server; 108 SNPs (288 mutations) were predicted to be deleterious out of 937 SNPs, Table (appendix). Deleterious SNPs were submitted to Polyphen-2 as query sequences in FASTA Format, 79 SNPs (202 mutations) were predicted to be probably damaging, the other 30 SNPs (53 mutations) were scored as possibly damaging, 101 SNPs (255 mutations) were predicted to be damaging by both servers (table 9 in the appendix). 21 SNPs (59 mutations) achieved high scores (TI= 0 I sift server and PSIC SD=1 by polyphen-2 software) and had been chosen for further analysis. Table (1)
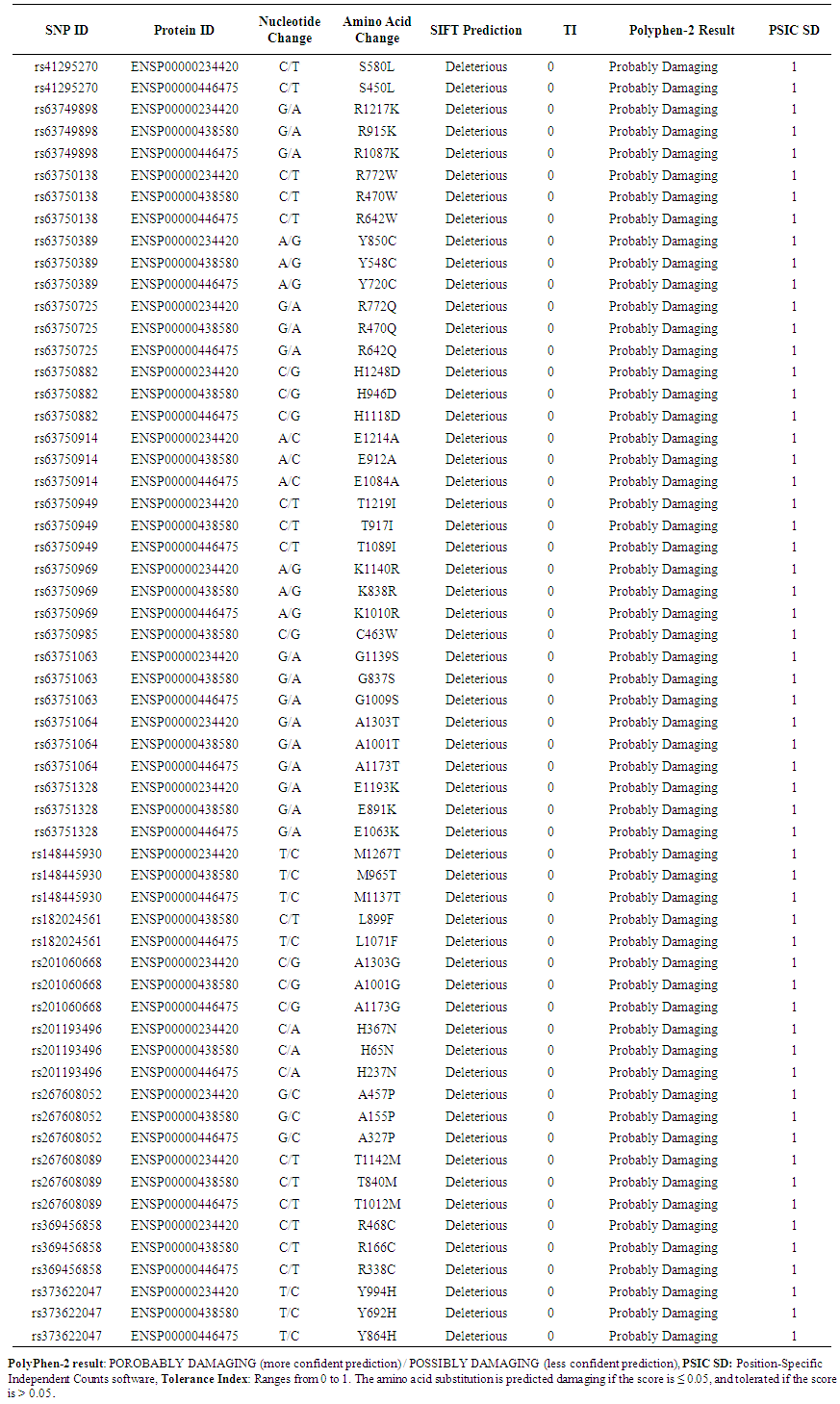 | Table 1. Shows highly damaging non synonymous SNPs predicted by SIFT and Polyphen2 |
3.4. Prediction of Harmful nsSNPs by Mutpred
- MutPred was used to determine the tolerance degree for each amino acid substitution on the basis of physio-chemical properties. The results obtained from MutPred server are shown in table (2). These results suggest that some nsSNPs may account for potential structural and functional changes in MSH6 protein.
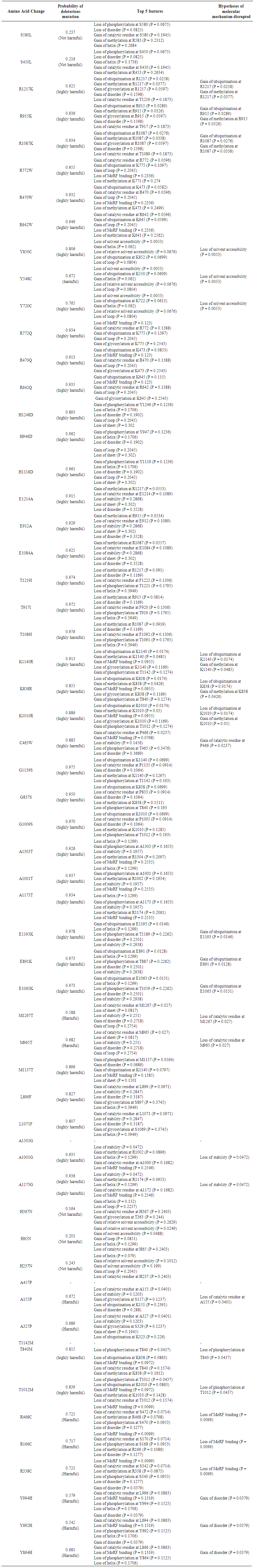 | Table 2. Prediction of the functional impact of nsSNPS on MSH6 protein by MutPred |
3.5. Identification of Disease Related nsSNPs by PhD-SNP and SNPs & GO
- PhD-SNP and SNPs & GO softwares were used to predict the association of SNPs with disease. According to PhD-SNP software, 19 SNPs (53 mutations) (R→K, R→W, Y→C, R→Q, H→D, E→A, T→I, K→R, C→W, G→S, A→T, E→K, M→T, L→F, A→G, Y→H, T→M and R→C) were disease related while 3 SNPs (6 mutations) (S→L, H→N, A→P) were predicted to be neutral polymorphisms. SNPs & GO predicted 7 SNPs (20 mutations) as neutral and 14 SNPs (39 mutations) (S→L, H→N, A→P, R→C, Y→H, Y→C and R→K) (R→W, R→Q, H→D, E→A, T→I, K→R, C→W, G→S, A→T, E→K, M→T, L→F, A→G, T→M and) were disease related. Table (3)
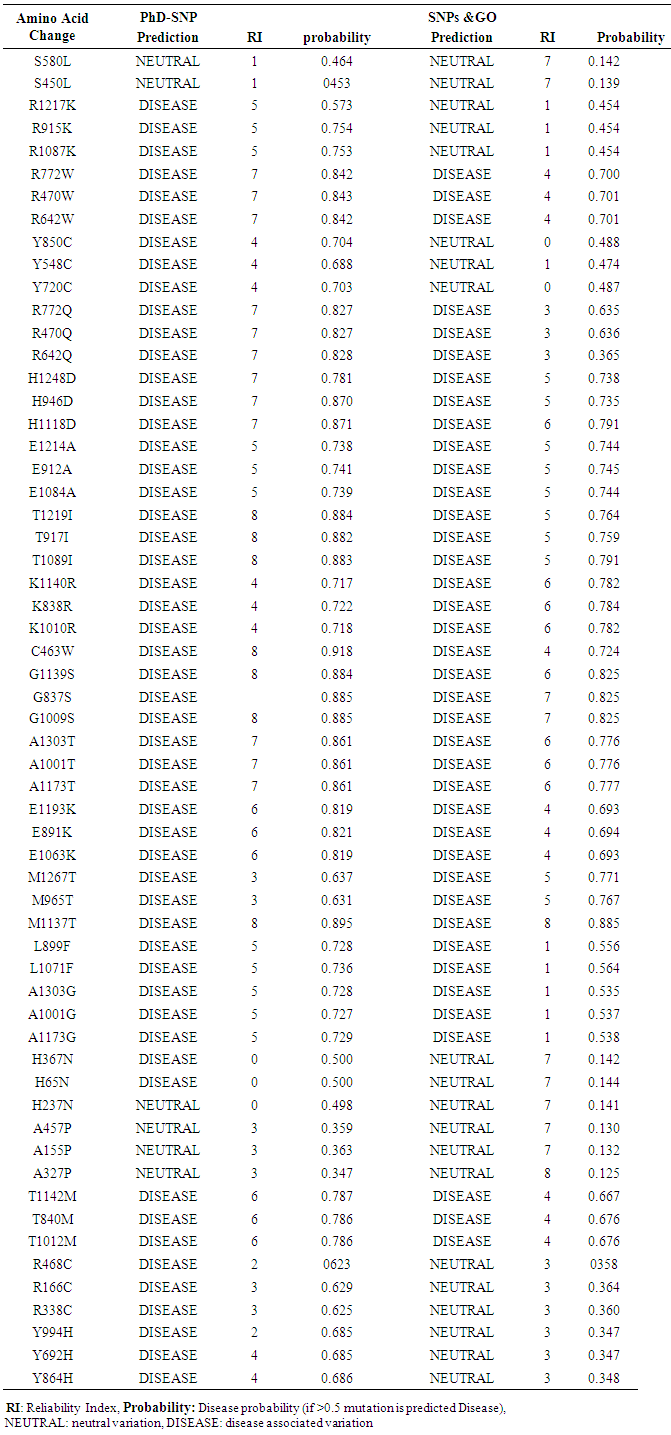 | Table 3. Shows prediction of disease related non synonymous SNPs by PhD-SNP and SNPs & GO |
3.6. Prediction of nsSNPs Impact on the Protein Stability by I-Mutant and MUpro
- In I-Mutant 3.0, the protein stability changed due to a single point mutation in 20 SNPs (56 mutations) in MSH6 gene (S→L, R→K, R→W, R→Q, H→D, E→A, T→I, K→R, C→W, G→S, A→T, E→K, M→T, L→F, A→G, H→N, A→P, T→M, R→C, Y→H) decrease effective stability of the protein and one SNP (3 mutations) (Y→C) was predicted to increase protein stability. In MUpro 6 SNPs (17mutations) (S→L, R→W, R→Q, H→D, K→R, T→M) were found to increase the protein stability and 15 SNPs (42mutations) were found to decrease protein stability (R→K, E→A, T→I, C→W, G→S, A→T, E→K, M→T, L→F, A→G, H→N, A→P, R→C, Y→H, Y→C). Table (4)
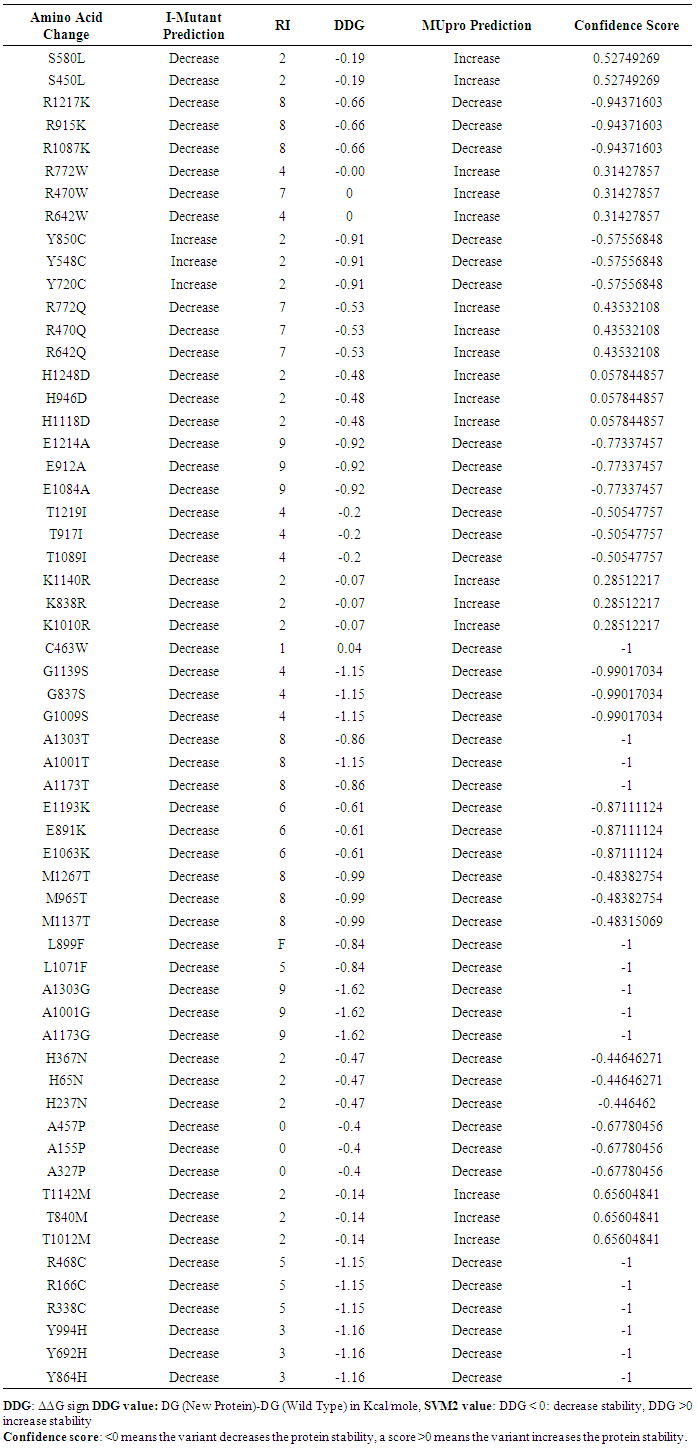 | Table 4. Shows the impact of nsSNPs on protein stability by I-mutant and Mupro |
3.7. Prediction of nsSNPs at the Protein Core and Interface
- ELASPIC server predicted 27 mutations in the core of MSH6 protein and 14 mutations were at the interface while 18 mutations were not found by the server. Table (5)
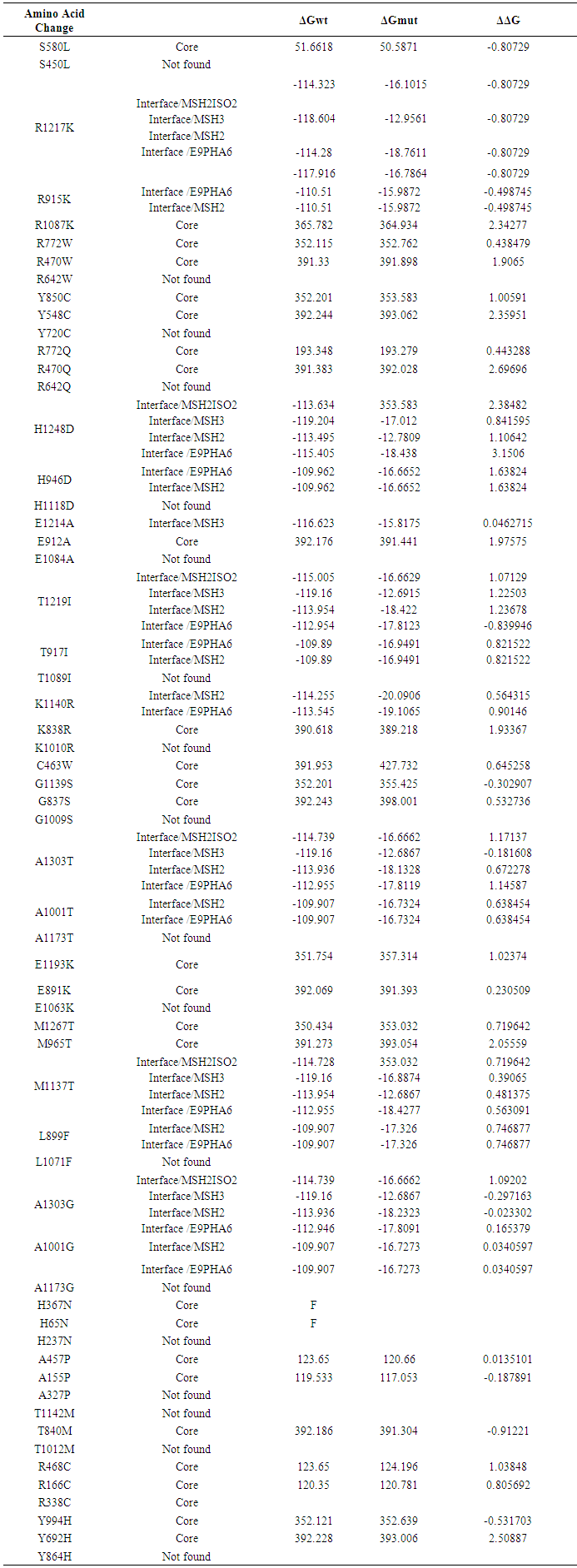 | Table 5. Prediction of stability effect on domain cores and domain-domains interfaces by ELASPIC |
3.8. Distribution of Mutations in MutS Domain by Mutation3D Server
- Five domain were detected in MSH6 protein; MutS I (PF01624), MutS II (PF05188), MutS III (PF05192), MutS V (PF00488), PWWP (PF00855) 54 mutations were located in a protein domain structure, 4 mutations were uncovered (H65N, H237N, A137P, R338C) and E1063 was located in the inter domain region. Those mutations in the domains regions are considered as higher risk mutations for MSH6 protein. The result is shown in figure 2.
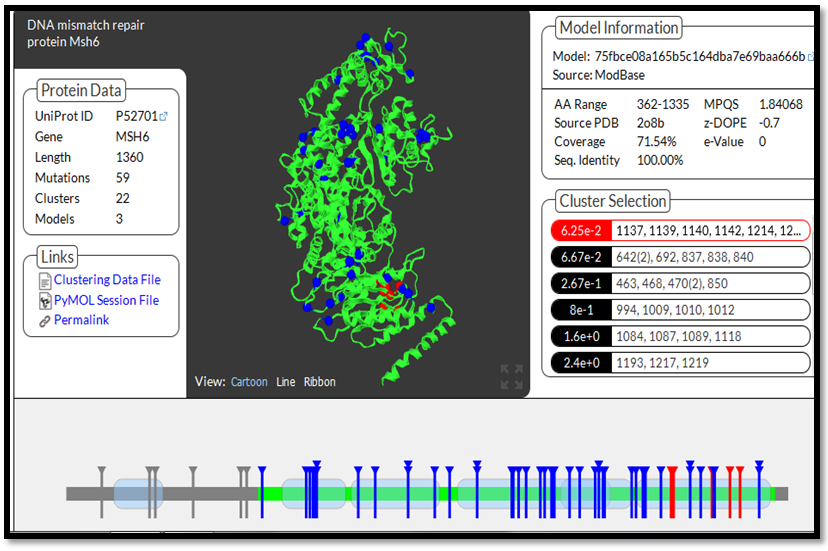 | Figure 2. MSH6 protein 3d structure and distribution of highly damaging mutations in protein's domains |
3.9. Protein 3d Modeling and Detection of Hydrogen Bonding and Clashes by UCSF Chimera
- UCSF Chimera v 1.8 was used to model the 3d structure of both wild and mutant residue and also to show hydrogen bonds, clashes and contacts of the mutant residue. Results are shown in figures 3-22.
 | Figure 3. rs41295270 (S580L): Wild residue (green color), mutant residue (red color), hydrogen bond (yellow lines) and clash (blue lines). One hydrogen bond interaction in wild and mutant residue, 4 clashes between mutant residue and ARG581 |
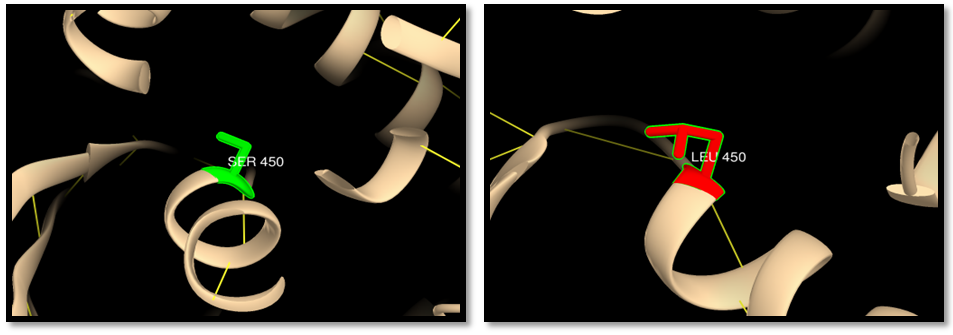 | Figure 4. rs41295270 (S450L) (MSH6 ISOFORM2): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow lines). One hydrogen bond interaction in wild and mutant residue |
 | Figure 5. rs63749898 (R1217K): wild residue (green color), mutant residue (red color) and clash (blue lines). 2 clashes between mutant residue and GLY 1218 |
 | Figure 6. rs63749898 (R9157K) (MSH6 ISOFORM4): wild residue (green color), mutant residue (red color), and clash (blue lines). 6 clashes of mutant residue with itself |
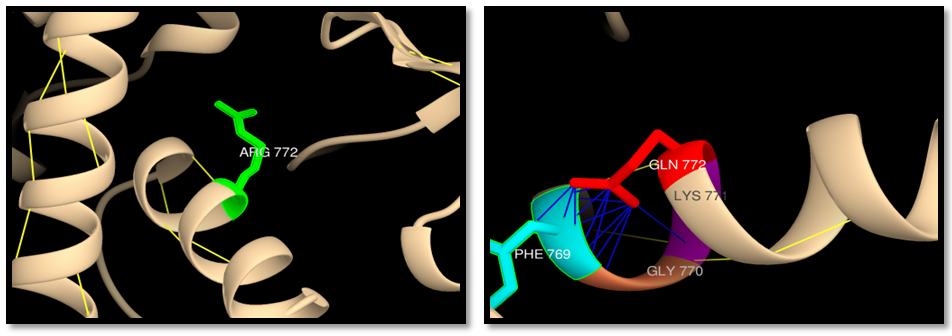 | Figure 7. rs63750725 (R772Q): wild residue (green color), mutant residue (red color), hydrogen bond (yellow lines) and clash (blue lines). One hydrogen bond interaction in wild and mutant residue, one clash between mutant residue and LYS771,one clash with GLY770 and 11 clashes with PHE769 |
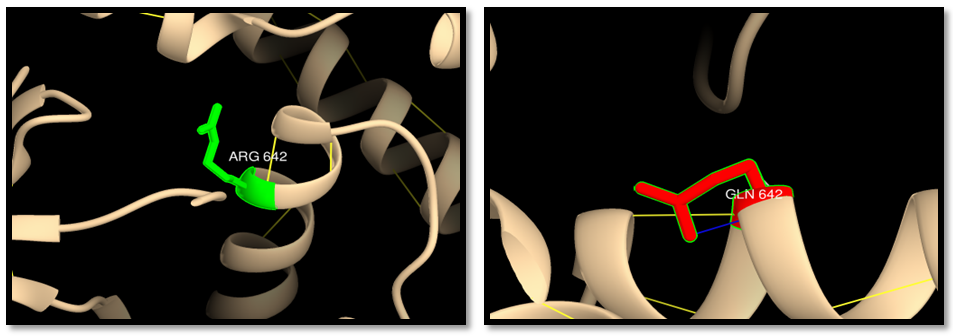 | Figure 8. rs63750725 (R642Q) (MSH6 ISOFORM3): wild residue (green color), mutant residue (red color), hydrogen bond (yellow lines) and clash (blue lines). One hydrogen bond interaction in wild and mutant residue, one clash of mutant residue with itself |
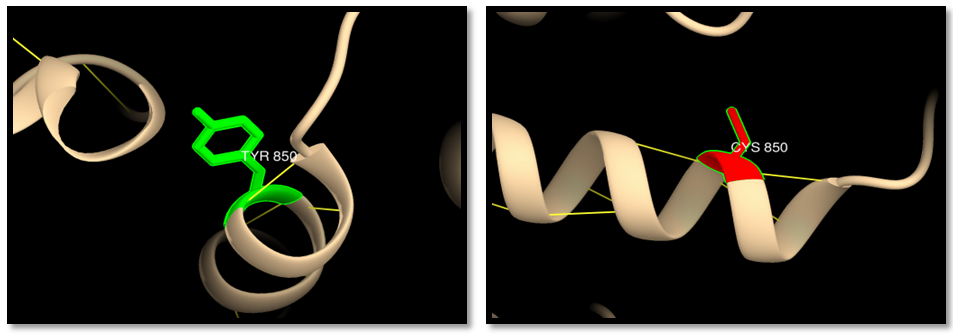 | Figure 9. rs63750389; (Y850C): wild residue (green color), mutant residue (red color), hydrogen bond (yellow lines) and clash (blue lines). 2 hydrogen bonds interaction in wild and mutant residue |
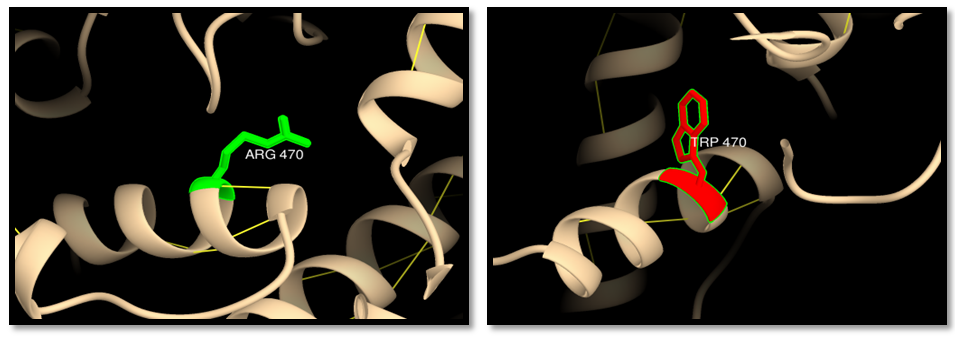 | Figure 10. rs63750138 (R470W) (MSH6 ISOFORM4): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow lines). One hydrogen bond interaction in wild and mutant residue |
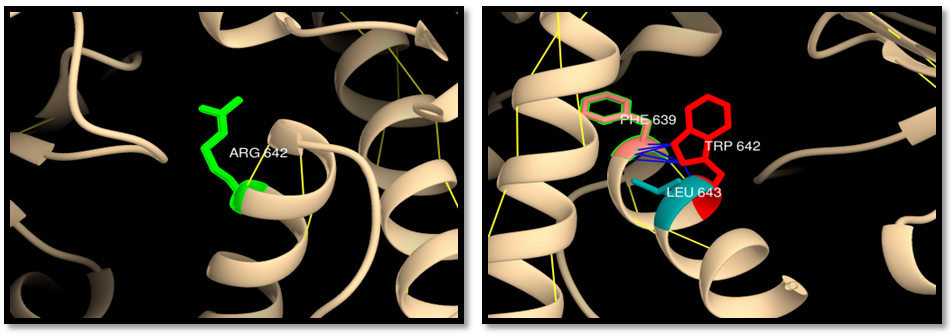 | Figure 11. rs63750138 (R642W) (MSH6 ISOFORM3): wild residue (green color), mutant residue (red color), hydrogen bond (yellow lines) and clash (blue line) One hydrogen bond in wild and mutant residue, one clash of mutant residue with LEU643 and 6 clashes with PHE639 |
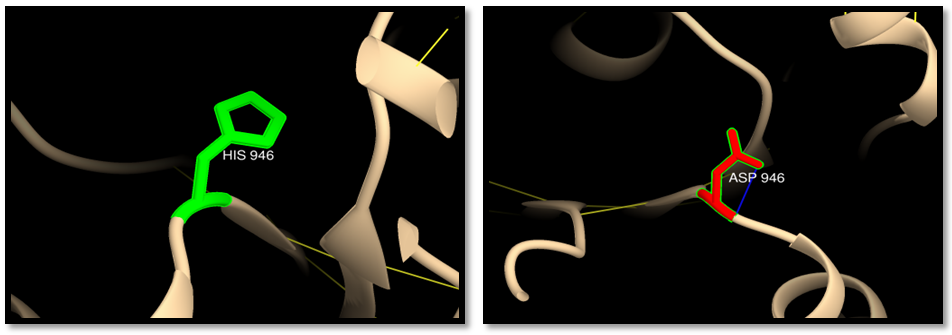 | Figure 12. rs63750882 (H946D) (MSH6 ISOFORM4): wild residue (green color), mutant residue (red color) and clash (blue line). One clash of mutant residue with itself |
 | Figure 13. rs63750914 (E1214A): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow lines). One hydrogen bond interaction in both wild and mutant residue |
 | Figure 14. rs63750969 (K1140R): wild residue (green color), mutant residue (red color) and clash (blue line). 4 clashes in mutant residue |
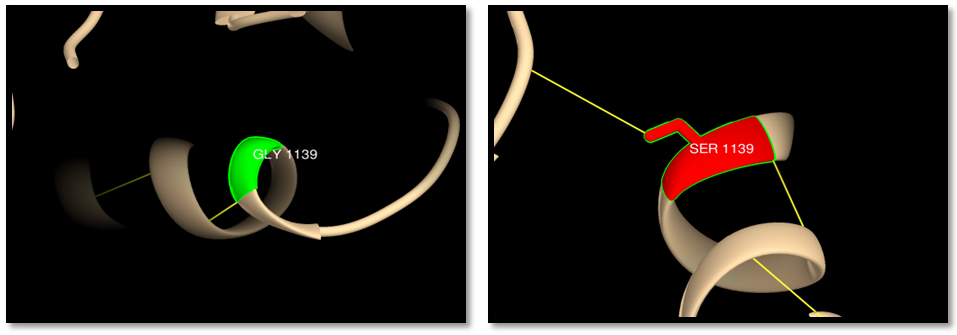 | Figure 15. rs63751063 (G1139S): wild residue (green color), mutant residue (red color), hydrogen bond (yellow line). One hydrogen bond in wild residue and 2 bonds in the mutant |
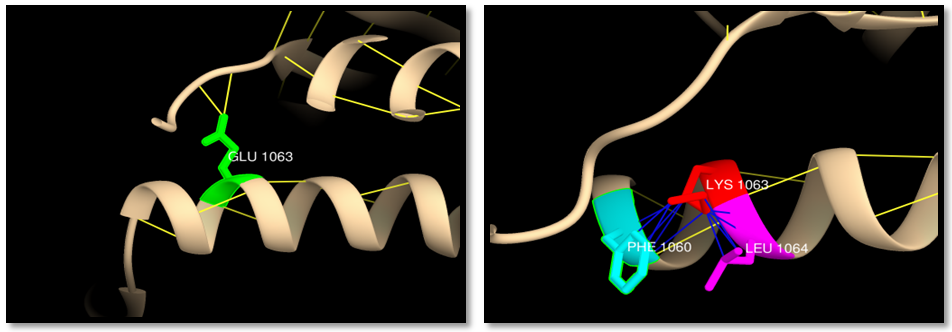 | Figure 16. rs63751328 (E1063K) (MSH6 ISOFORM3) wild residue (green color), mutant residue (red color), clash (blue line) and hydrogen bond (yellow line). 3 hydrogen bonds in wild residue and 2 in mutant,6 clashes of mutant residue with PHE1060 and 5 clashes with LEU1064 |
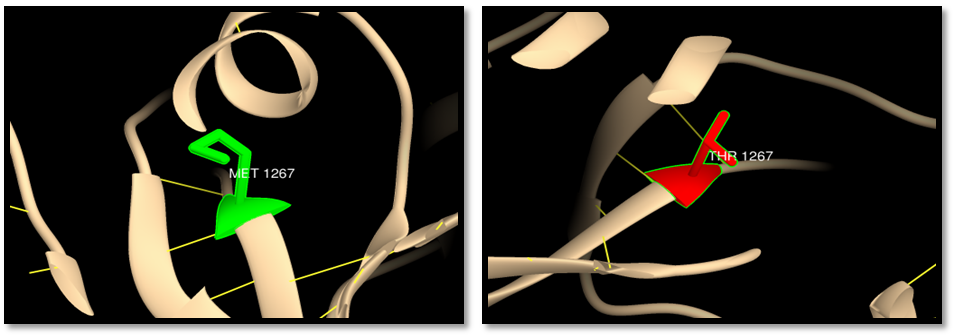 | Figure 17. rs148445930 (M1267T): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow line). One hydrogen bond in both wild and mutant residue |
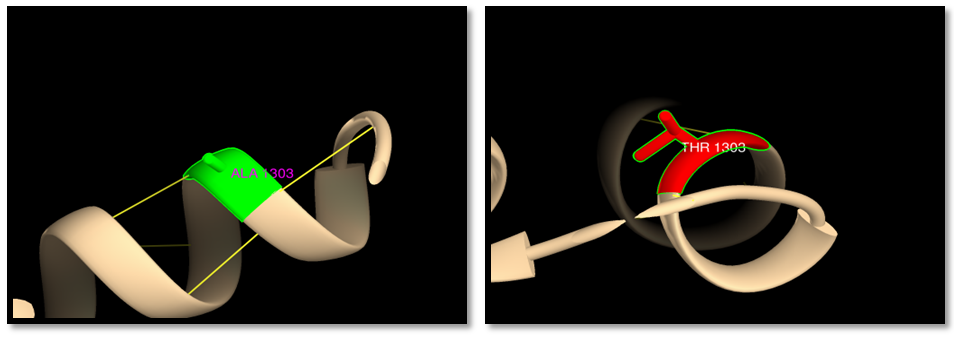 | Figure 18. rs201060668 (A1303G): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow line). One hydrogen bond in both wild and mutant residue |
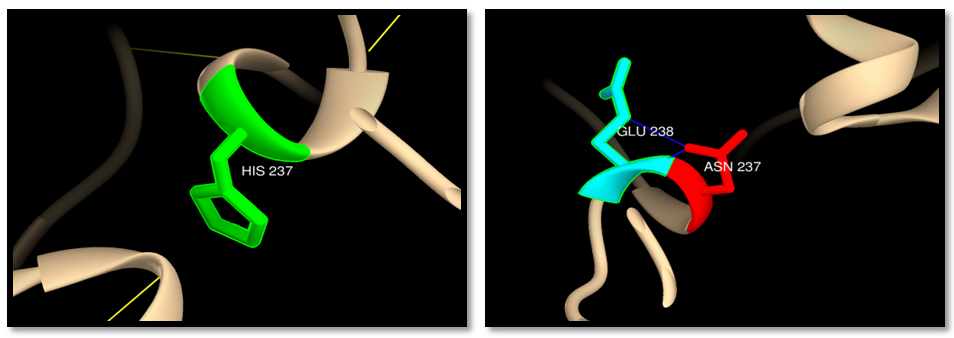 | Figure 19. rs201193496 (H237N): wild residue (green color), mutant residue (red color) and clash (blue line). 2 clashes between mutant residue and GLU238 |
 | Figure 20. rs267608089 (T1142M): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow line). Three hydrogen bonds in wild residue and two bonds in mutant residue |
 | Figure 21. rs369456858 (R468C): wild residue (green color), mutant residue (red color) and hydrogen bond (yellow line). One hydrogen bond in wild residue and two bonds in mutant residue |
 | Figure 22. rs373622047 (Y864H) (MSH6 ISOFORM3): wild residue (green color), mutant residue (red color), hydrogen bond (yellow line) and clash (blue line). One hydrogen bond in wild and mutant residue, one clash in the mutant residue |
3.10. Influence of SNPS at the 3UTR on miR Binding Sites by PolymiRTS Database
- Regarding the analysis of SNPs at the 3`UTR region using PolymiRTS database, out of 63 SNPS 2 functional SNPs were predicted to affect miRSite; rs200412142 was found to has 2 alleles (A and G) contained 2 (D) and 1(C) functional classes on 3 miRSite while rs184571821 SNP had 2 alleles (C) contained 3(C) functional class had 3 miRSites; (D) functional class disrupts a conserved miRNA site while (C) is a target binding site that can create a new microRNA site, Table (6).
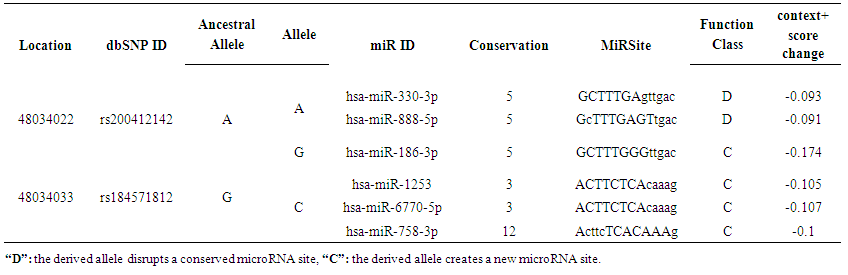 | Table 6. Shows the SNPs predicted by PolymiRTS to induce disruption or creation of mirRNA binding site |
4. Discussion
- The human MSH6 protein was first reported in 1995 as G/T mismatch Binding Protein (GTBP), binding partner of hMSH2 to form the MutSα complex. The hMSH6 gene product is a 160 kDa protein that is unstable without heterodimerization with hMSH2, and consequently utilizes 80%–90% of available hMSH2. MSH2 and MSH6 share five similar domains, but with sufficient differences to give MSH6 several distinct functions. MSH6 also has a unique N-terminal disordered domain that is absent in its MSH2 partner. The hMutSα heterodimer binds to DNA mispairs and short insertion deletion loops (IDLs) [13, 60-64]. Mutations of MSH6 gene had been reported to be associated with many cancers but mainly with HNPCC syndrome, and to date the exact mechanism of how these mutations promote tumor genesis remains controversial, in the present study we presented a computational analysis for the reported MSH6 SNPs using several public softwares and databases in an attempt to understand how do these mutations affect the protein structure and function and hence promote a disease. To sort out tolerant from intolerant nsSNPs ten different prediction algorithm were used; SIFT, Polyphen, PHD-SNPs, SNP&GO, I-Mutant, MUpro, Mutpred, ELASPIC, Mutation 3D and USCF Chimera. GENEMANIA was used to investigate MSH6 gene interactions and its role in net works. PolymiRTs database was used to analyze the impact of SNPs at the 3UTR on microRNAs binding sites. Our findings showed 108 nsSNPs (288 mutations) out of 937 nsSNPs were predicted to be deleterious by SIFT, and 21 nsSNPs (59 mutations) were predicted to be highly damaging by SIFT and Polyphen (table 1,9) and those were selected for further analysis by other insilico tools. 19 nsSNPs (53 mutations) were predicted to be disease related by PHD-SNPs while SNP&GO predicted 14 SNPs (39mutations) to be disease related (table 3). The differences in prediction capabilities refer to the fact that every prediction algorithm uses different sets of sequences and alignments. Mutpred was used to determine the tolerance degree for each amino acid substitution on the basis of physio-chemical properties, the results showed 41 mutations were highly harmful (table 2), Gain of ubiquitination and methylation for the mutation (R→K) at positions 1217,915 and 1087, Loss of solvent accessibility for the mutation (Y→C) at positions 850, 548 and 720, loss of ubiquitination and gain of methylation for the mutation (K→R) at positions 1140, K838 and 1010, Gain of catalytic residue at P466 for the mutation C463W, Gain of ubiquitination for the mutation (E→K) at positions 1193, 891 and 1063, Loss of catalytic residue for the mutation at positions (M→T) 1267 and 965, loss of stability for the mutation (A→G) at positions 1001 and 1173, Loss of catalytic residue for the mutation (A→P) at position 155, Loss of phosphorylation for the mutation (M→T) at positions 1012 and 840, Loss of MoRF binding for the mutation (R→C) at positions 468, 166 and 388, Gain of disorder for the mutation (Y→H) at positions 994, 692 and 864. These results indicate that some nsSNPs account for potential structural and functional changes in MSH6 protein. For further confirmation nsSNPs were submitted to I-Mutant and MUpro and the findings were; In I-Mutant 3.0, 20 nsSNPs (56 mutations) (S→L, R→K, R→W, R→Q, H→D, E→A, T→I, K→R, C→W, G→S, A→T, E→K, M→T, L→F, A→G, H→N, A→P, T→M, R→C, Y→H) decreased the effective stability of the protein while in MUpro 15 nsSNPs (42mutations) were found to decrease the protein stability (R→K, E→A, T→I, C→W, G→S, A→T, E→K, M→T, L→F, A→G, H→N, A→P, R→C, Y→H, Y→C). ELASPIC server was used to classify nsSNPS at the core or interface of the protein. The results showed 26 mutations (S580L, R1087K, R770W, Y850C, G1139S, E1193K, M1267T, H367N, A457P, R468C, R338C, Y994H, T1142M, R470W, Y548C, R470Q, E912A, K838R, C463W, G837S, E891K, R166C, T840M, R772Q, M965T, Y692H ) were in the core of MSH6 protein and 14 mutations (R1217K, H1248D, E1214A, T1219I, K1140R, A1303T, M1137T, A1303G, R915K, H946D, T917I, A1001T, L899F, A1001G) were at the interface. The “core” residues are defined as residues which are exposed in the monomeric protein but buried in the protein complex. Core residues are typically hydrophobic with a composition strongly divergent from the composition of the remainder of the protein surface [65]. Core residues supply the bulk of the energy driving association by hydrophobic interactions [66]. The hydrophobic interactions within the complex cause the core region to become tightly packed upon complex association with little room for conformational variability. For these reasons, the core residues are strongly conserved during evolution [67] and mutations in this region are usually more strongly unfavorable when compared to mutations at the periphery of the interface. In cancer, 3D location of mutations at an interface has served as evidence that protein interactions may be important for metastasis site determination. Mutation3D was used to investigate the distribution of nsSNPs in MutS domains. NsSNPs were distributed in 5 domains of MSH6 protein, MutS I (R470W, R470Q, S540L, C463W, R468C, A 457P), MutS II (R642W, R642Q, S58L,Y548C,Y692H), MutS III (H946D, R915K, Y850C, R772W, R772Q, E912A, K1010R,A100T, M965T, T917I, L899F, E891K, K838R,T1012M, A100G, Y994H, Y864H, T840M), MutS V (H1248D, E1214A, H1118D, R1087K, R1217K, A1303T, M1267T, T1219I, E1193K, A1173T, M1137T, K1140R, E1084A, T1089I, A 1303G, A1173G, T1142M), PWWP (R166C, A155P) while H65N, H237N, A327P and R338C were uncovered mutations, E1063K and L1071F were located at inter domain regions. Mutations within the protein domains are considered high risk mutations which may lead to disturbance or loss of the protein function. 3D structure of MSH6 protein, H bond and clash was shown between wild type and mutant using the visualized chimera program. G1139S, E1193K, E891K, E1063K, T1142M, R468C, Y864H had difference in number of hydrogen bonds between wild and mutant residue indicating that these mutation will disturb the stability of the protein. Cash was detected in the mutant residue (S580L, R1217K, R915K, R772Q, R642Q, R642W, H946D, K1140R, E1063K, H237N, Y864H) indicating a change in the environment of the molecule and hence a change in the structure and function of the protein. rs41295270 (S580L and S450L): The residue is located in the core of the protein and mutation of this residue can disturb interactions with other molecules or other parts of the protein. The mutant residue is located near a highly conserved position. The wild-type and mutant amino acid differ in size. The mutant residue is bigger than the wild-type and more hydrophobic, the size difference between wild-type and mutant residue makes that the new residue is not in the correct position to make the same hydrogen bond as the original wild-type residue did. Also difference in hydrophobicity will affect hydrogen bond formation. GSK3 phosphorylation site (MOD_GSK3_1) motif predicted to be at this position is damaged by the mutation, only serine, threonine and tyrosine residues can be phosphorylated and mutation into another residue type will disturb this modification. The mutant residue has 4 clashes with ARG 581 which will change the environment of the molecule. The mutation will cause Loss of disorder (P=0.0825), gain of catalytic residue at S580 (P=0.1945), gain of methylation at R583 (P=0.2312), gain of helix (P = 0.2684), gain of catalytic residue at S450 (P = 0.1945) and gain of methylation at R453 (P = 0.2634).rs63749898 (R1217K, R915K and R1087K): The mutant residue is located near a highly conserved position. The mutant residue is smaller than the wild-type and this may cause a possible loss of external interactions. The wild-type residue forms a hydrogen bond with: glutamic acid at position 1214, glycine at position 1216, the size difference between wild-type and mutant residue makes that the new residue is not in the correct position to make the same hydrogen bond as the original wild-type residue did, also difference in hydrophobicity will affect hydrogen bond formation. PKA Phosphorylation site (MOD_PKA_2) motif predicted to be at this position is damaged by the mutation. The mutant residue has 2 clashes with GLY1218. The mutation will cause gain of disorder (P = 0.1598).rs63750138 (R772W, R470W and R642W): The mutant residue is bigger and more hydrophobic than the wild residue the charge of the buried wild-type (positive) residue is lost by this mutation. The wild-type and mutant amino acids differ in size. The mutant residue is bigger than the wild-type which is buried in the core of the protein but the mutant residue probably will not fit. The mutation will cause loss of hydrogen bonds in the core of the protein and as a result disturbs correct folding. The mutant residue has 1 clash with LEU643 and 6 clashes with PHE639. The mutation will cause gain of loop (P=0.2045) and loss of MoRF binding (P = 0.2336). This mutation matches a previously described variant (VAR_043958) of Hereditary non-polyposis colorectal cancer 5 (HNPCC5) [MIM: 614350] which is annotated with the severity of the disease.rs63750389 (Y850C, Y548C and Y720C). The wild-type residue forms a hydrogen bond with: serine at positions 564,262,434 and valine at positions 594,292,464. The wild-type residue forms a salt bridge with: serine at position 564,262,434, valine at position 592,290,464 glutamic acid at position 847,545,717, threonine at position 849,547,719. The wild-type residue is predicted (by KMAD) to be a phosphorylation site and only serine, threonine and tyrosine residues can be phosphorylated and mutation into another residue type will disturb this modification. The mutant residue is smaller than the wild-type residue and this difference will cause an empty space in the core of the protein. The mutation will cause loss of hydrogen bonds in the core of the protein and as a result disturb correct folding. This mutation matches a previously described variant (VAR_012963) associated with HNPCC5 and Colorectal cancer, reported by Ying et al (1999) in Chinese patients with hereditary non polyposis colorectal cancer [68].rs63750725 (R642W, R772W and R470W). There is a difference in charge and size between the wild-type (positively charged) and mutant (smaller and neutral) amino acid. The wild-type residue forms a hydrogen bond with: glutamic acid at position 1084, 1214 and glycine at position 1086, 1216. The size difference between wild-type and mutant residue makes that the new residue is not in the correct position to make the same hydrogen bond as the original wild-type residue did. The mutant residue has 1 clash with LEU643 and PHE639. The mutation will cause gain of loop (P=0.2045) and loss of MoRF binding (P = 0.2336).rs63750882 (H1248D, H946D and H1118D). There is a difference in charge between the wild-type (neutral) and mutant (negatively charged) amino acid and this can lead to protein folding problems. The mutant residue is smaller than the wild-type residue and this will cause an empty space in the core of the protein. According to the PISA-database, the mutated residue is involved in a multimer's contact; the mutation introduces a smaller residue at this position. The new residue might be too small to make multimer contacts. The motif NEK2 phosphorylation site (MOD_NEK2_1) is damaged by the mutation. The mutant residue at position 946 has 1 clash. The mutation will cause loss of helix (P = 0.1706), loss of disorder (P = 0.1902), gain of loop (P = 0.2045) and loss of sheet (P = 0.302). rs63750914 (E1214A, E912A and E1084A). The wild-type residue was negatively charged while the mutant residue is neutral and smaller than the wild-type. The mutation will cause loss of hydrogen bonds in the core of the protein and as a result disturbs correct folding. The wild-type residue forms a hydrogen bond with: arginine at positions 772,470,642. The size difference between wild-type and mutant residue makes that the new residue is not in the correct position to make the same hydrogen bond as the original wild-type residue did. The mutation will cause loss of stability (P = 0.2868), loss of sheet (P = 0.302) and Loss of disorder (P = 0.3528).rs63750949 (T1219I, T917I, and T1089I). There is a difference in size and hydrophobicity between the wild-type and mutant (bigger and more hydrophobic) amino acid. The wild-type residue is predicted (by KMAD) to be a phosphorylation site and mutation into another residue type will disturb this modification. In both the PDB-file and in the PISA-assembly, this residue was found to be involved in a multimer's contact. This is a strong indication that the residue is indeed in contact with other proteins. The mutation introduces a bigger residue at these positions; this can disturb the multimeric interactions. A more hydrophobic residue is introduced and any hydrogen bond that could be made by the wild-type residue to other monomers will be lost and affect the multimeric contacts. The mutation will cause loss of helix (P = 0.3949).rs63750969 (K1140R, K838R and K1010R): The wild-type residue has interactions with a ligand annotated as ADP The difference in properties between wild-type and mutation can easily cause loss of interactions with the ligand. Because ligand binding is often important for the protein's function, this function might be disturbed by this mutation. According to the PISA-database, the mutated residue is involved in a multimer contact. The mutations introduce bigger residues at these positions which can disturb the multimeric interactions. The wild-type residue is located in a region annotated in UniProt to form an α-helix. The mutations convert the wild-type residue in to residue that do not prefer α-helices as secondary structure. rs63750985 (C463W): The mutant residue is bigger than the wild-type which is buried in the core of the protein so the mutant residue probably will not fit. The mutantion will cause gain of catalytic residue at P466 (P = 0.0237), gain of MoRF binding (P = 0.0768), loss of stability (P = 0.1456), loss of phosphorylation at T465 (P = 0.3476) and loss of disorder (P = 0.3669).rs63751063 (G1139S, G837S and G1009S): The mutant residue is bigger than the wild-type residue. The mutatnt residue is not in direct contact with a ligand, however, the mutation could affect the local stability which in turn could affect the ligand-contacts made by one of the neighboring residues. These differences in properties between wild-type and mutant residue can easily cause loss of interactions with the nucleotide ("ATP"). This can directly affect the function of the protein. The wild-type residue forms a salt bridge with: glycine at positions 1138,836 and 1008.The wild-type residue forms a salt bridge with: glycine at these positions. The torsion angles for this residue are unusual. Only glycine is flexible enough to make these torsion angles, so mutation into another residue will force the local backbone into an incorrect conformation and will disturb the local structure. The mutation will cause loss of helix (P = 0.1299), loss of disorder (P = 0.2501) and loss of stability (P = 0.2938).rs63751064 (A1303T, A1001T and A1173T): There is a difference in hydrophobicity and size between the wild-type (more hydrophobic) and mutant (bigger) amino acid. The wild-type residue is located in a region annotated in UniProt to form an α-helix. The mutation converts the wild-type residue in a residue that does not prefer α-helices as secondary structure. WDR5 WD40 repeat (blade 5, 6)-binding ligand (LIG_WD40_WDR5_VDV_2) motif is damaged by the mutation. The mutation will cause loss of helix (P = 0.1299) and loss of stability (P = 0.1957) Loss of MoRF binding (P = 0.2535).rs63751328 (E1193K, E891K and E1063K). There is a difference in charge and size between the wild-type (negatively charged) and mutant (positively charged and bigger) amino acid. The charge of the buried wild-type residue is reversed by this mutation; this can cause repulsion between residues in the protein core. The mutant residue is bigger than the wild-type residue which buried in the core of the protein so the mutant residue probably will not fit. The mutant residue has 6 clashes with PHE 1060 and 5 clashes with LEU1063. The mutation will cause loss of helix (P = 0.1299), loss of disorder (P = 0.2501) and loss of stability (P = 0.2938). This mutation matches a previously described variant (VAR_043970), found in an endometrial cancer sample; displays marked impairment of heterodimerization with MSH2 and of in vitro mismatch repair capacity. rs148445930 (M1267T, M965T and M1137T): The mutant residue is smaller than the wild-type residue and this might cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs and the mutation might cause loss of hydrophobic interactions with other molecules on the surface of the protein. The mutation will cause loss of sheet (P = 0.0817), loss of stability (P = 0.251), gain of disorder (P = 0.2718) and gain of loop (P = 0.2754).rs182024561 (L899F and L1071F): The wild-type residue is highly conserved. The mutant residue is bigger than the wild-type residue is buried in the core of the protein so the mutant residue probably will not fit. The mutation will cause loss of stability (P = 0.2847), loss of disorder (P = 0.3187) and loss of helix (P = 0.3949).rs201060668 (A1303G, A1001G and A1173G): The mutation introduces a glycine at these positions. Glycine is very flexible and can disturb the required rigidity of the protein at these positions. Mutant residue is smaller than the wild-type residue and this will cause a possible loss of external interactions. There is difference in hydrophobicity between the wild-type (more hydrophobic) and mutant residue and this might cause loss of hydrophobic interactions with other molecules on the surface of the protein. WDR5 WD40 repeat (blade5,6)-binding ligand (LIG_WD40_WDR5_VDV_2) is damaged by the mutation. The mutation will cause loss of stability (P = 0.0472), loss of helix A1000 (P = 0.1682) and loss of MoRF binding (P = 0.2546).rs201193496 (H367N, H65N and H237N): The mutant residue is smaller than the wild-type residue and this will cause an empty space in the core of the protein. The new residue might be too small to make multimeric contacts. The residue is buried in the core of a domain. The differences between the wild-type and mutant residue might disturb the core structure of this domain. The mutant residue has 2 clashes with GLU238. The mutation will cause gain of relative solvent accessibility (P = 0.0249), gain of solvent accessibility (P = 0.0488), gain of loop (P = 0.0851) and loss of helix (P = 0.1299).rs267608052 (A457P, A155P and A327P): There is difference in size between the wild-type and mutant residue. The mutant residue is bigger than the wild-type residue which buried in the core of the protein so the mutant residue probably will not fit. The wild-type residue forms a hydrogen bond with: aspartic acid. The size difference between them makes that the new residue is not in the correct position to make the same hydrogen bond as the original wild-type residue did. The mutation will cause Loss of stability (P = 0.1203), gain of sheet (P = 0.1945).rs267608089 (T1142M, T840M and T1012M): The mutant residue is bigger than the wild-type residue which is located on the surface of the protein, mutation of this residue can disturb interactions with other molecules or other parts of the protein. The mutant residue is more hydrophobic than the wild residue so any hydrogen bond that could be made by the wild-type residue to other monomers will be lost and affect the multimeric contacts. The difference in properties between wild-type and mutation can easily cause loss of interactions with the ligand. Because ligand binding is often important for the protein's function, this function might be disturbed by this mutation. The mutation may cause gain of MoRF binding (P=0.0972).rs369456858 (468, 166 and 338): The mutant residue is smaller than the wild-type residue and this will cause a possible loss of external interactions. The charge of the wild-type (positive) residue is lost by this mutation which can cause loss of interactions with other molecules. The mutation may cause loss of MoRF binding (P = 0.0099) and loss of disorder (P = 0.1277).rs373622047 (Y994H, Y692H and Y864H): The mutant residue is smaller than the wild-type residue and this will cause possible loss of external interactions. The wild-type residue is predicted to be located in its preferred secondary structure, a β-strand. The mutant residue prefers to be in another secondary structure; therefore the local conformation will be slightly destabilized. There is difference in the hydrophobicity between the wild-type and mutant residue. The mutation might cause loss of hydrophobic interactions with other molecules on the surface of the protein. The mutation might cause gain of disorder (P = 0.0379), loss of MoRF binding (P = 0.1319) and loss of helix (P = 0.1706).MicroRNAs (miRNAs) are negative gene regulators acting at the 3'UTR level, modulating the translation of cancer-related genes. Single-nucleotide polymorphisms (SNPs) within the 3'UTRs could impact the miRNA-dependent gene regulation either by weakening or by reinforcing the binding sites. Thus, the alteration of the normal regulation of a given gene could affect the individual's risk of cancer [69]. It is helpful to predict which of the many SNPs could really impact the regulation of a target gene. We used polymiRTS database to predict the impact of SNPs at the 3UTR on micro RNAs binding sites and the results showed 2SNPs out of 63 at the 3UTR disrupt microRNAs binding sites and hence affect the gene expression. rs200412142 was found to has 2 alleles (A and G) contained 2 (D) and 1(C) functional classes on 3 miRSite while rs184571821 SNP had 2 alleles (C) contained 3(C) functional class had 3 miRSites; (D) functional class disrupts a conserved miRNA sites while (C) is a target binding site that can create a new microRNA sites. Recently micro RNAs have became the top important objects for revolutionary molecular research and many experiments had been conducted to assess the potentiality of using micrRNAs as targeted treatments for many cancers.MSH6 is known of its DNA repairing function, yet GENMANIA revealed a possible role in B cell activation involved in immune response, production of molecular mediator of immune response, immunoglobulin production and somatic recombination of immunoglobulin genes involved in immune response, suggesting vital roles in immune system which might be an interesting area for future research.
5. Conclusions
- 21 nsSNPS were predicted by different softwares to be the most damaging mutations for MSH6 protein altering physiochemical properties of the protein; size, charge and hydrophobicity leading to loss or disturbance of the protein internal and external interactions and eventually loss of the protein's function. 14 nsSNPs (R1217K, H1248D, E1214A, T1219I, K1140R, A1303T, M1137T, A1303G, R915K, H946D, T917I, A1001T, L899F, A1001G) were located at the interface of the MSH6 protein interfering with its relation with MSH2ISO2, MSH3, MSH2 and E9PHA6. Interactions of MSH6 with these proteins are critical for its MMR function and any structural alterations that interfere or harm these interactions would probably increase susceptibility to tumors formation and progression. 2 SNPs at the 3UTR; rs200412142 and rs184571821 introduced a change in the micro RNA binding site at the 3UT which might result in deregulation of the gene function.
ACKNOWLEDGMENTS
- We thank the National Center for Neurological Sciences (NCNS) for the technical support.
Appendix
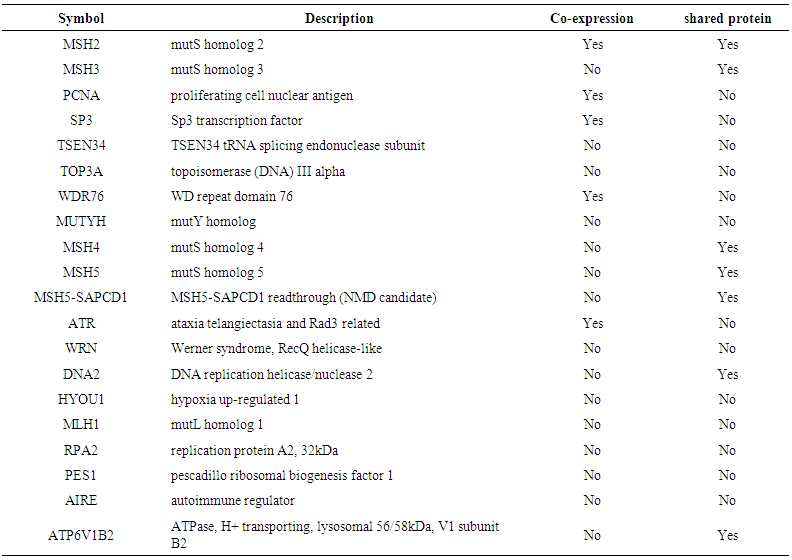 | Table 7. Shows the genes co-expressed and sharing a domain with MSH6 |
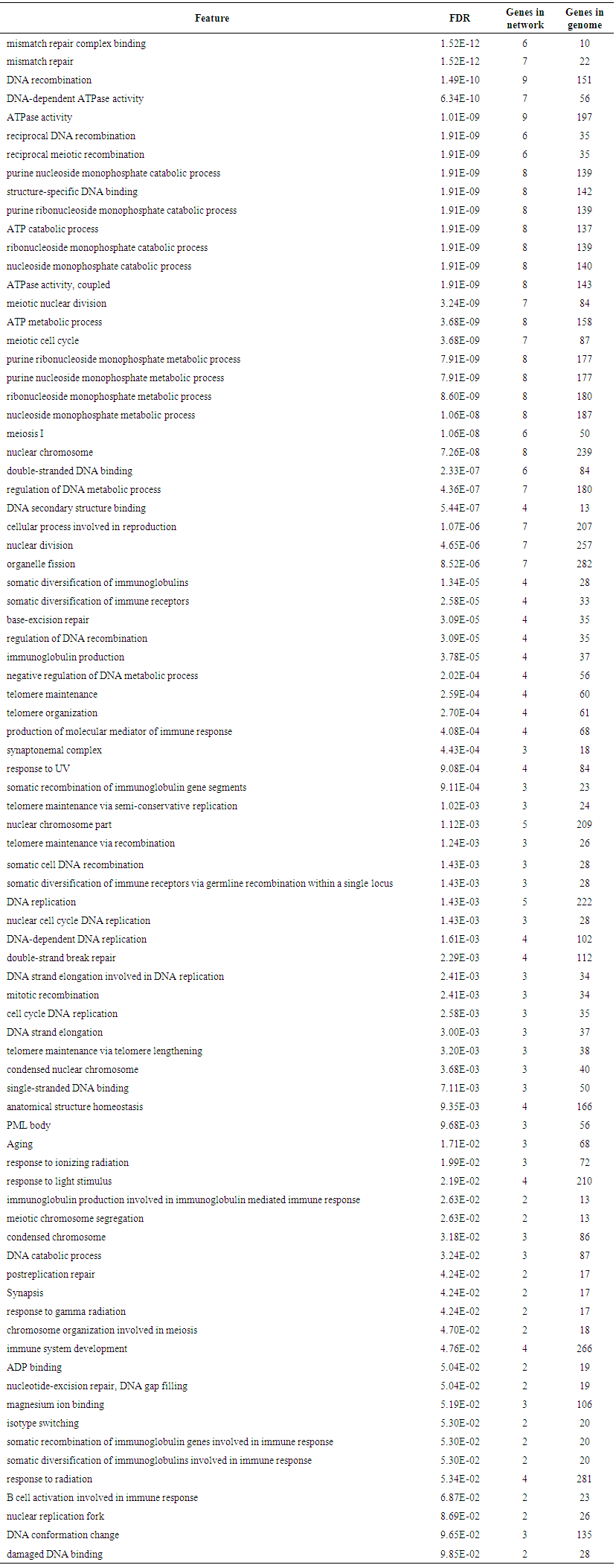 | Table 8. Shows MSH6 functions and its appearance in network and genome |
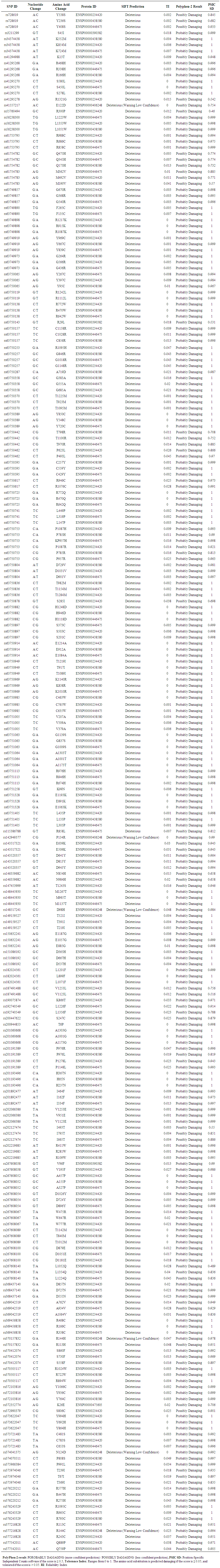 | Table 9. Shows results of deleterious SNPs predicted by SIFT, Polyphen-2 |