-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2015; 5(1): 16-19
doi:10.5923/j.bioinformatics.20150501.03
Assemblies of Wheat EST Sequences and Annotation of Affymetrix Consensus Sequences of Wheat Transcriptome
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLShailesh Sharma
National Agri-Food Biotechnology Institute, Department of Biotechnology, Government of India, Mohali, India
Correspondence to: Shailesh Sharma , National Agri-Food Biotechnology Institute, Department of Biotechnology, Government of India, Mohali, India.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Wheat is an important crop in the world, having the most challenging genomes. Triticum aestivum commonly known as bread wheat is a hexaploid, with three complete genomes termed as A, B and D in the nucleus of each cell. Each of these genomes is almost twice of the human genome and consists of around 6,000 million letters. Presently complete wheat genome sequence is not present. For this reason wheat genome research groups are using different genome assembly approaches and tools. In this paper, we report assembly of wheat ESTs by two different freewares CAP3 [1] and Trinity [2]. The second is an assembly of 61115 Affymetrix consensus sequences representing 42 chromosomes of bread wheat with EST sequences for increasing their length and annotation on the basis of sequence similarity with A, B and D gene models. We report the assembled contig sequences by two different freewares CAP3 [1] and Trinity [2] with their address in the genome which was a lacking information earlier. Trinity [2] was found in the better then CAP3 [1] program because Trinity contigs in comparison to the CAP3 contigs are mapped over gene models much better.
Keywords: EST, Triticum aestivum, Python, CAP3, Trinity
Cite this paper: Shailesh Sharma , Assemblies of Wheat EST Sequences and Annotation of Affymetrix Consensus Sequences of Wheat Transcriptome, American Journal of Bioinformatics Research, Vol. 5 No. 1, 2015, pp. 16-19. doi: 10.5923/j.bioinformatics.20150501.03.
Article Outline
1. Introduction
- The genome sequence of Triticum aestivum also known as bread wheat, holds the key to its genetic improvements. This will allow researchers to help growers to meet the increasing demands for high quality food and feed produced in an ever changing environment conditions. Globally, wheat is a major source of protein in the human diet. Affymetrix Wheat Genome array [www.affymetrix.com] provides researchers a single array covering all 42 chromosomes, and is widely used for gene expression studies. This is useful for gaining a better understanding of the wheat genome studies on how to develop innovative methods to feed a growing world population. Presently complete genome sequence of bread wheat is lacking. EST sequences available in public domain require efficient softwares for assembly. In the present study, we performed assembly of wheat EST sequences by two different freewares using default parameters and we tried to locate them all on available A, B and D gene models of Triticum aestivum.
2. Material and Methods
- Presently wheat genome researchers are having 99386 coding sequences (CDS) of all high-confidence (HCS) gene models [3] having a home on the International Wheat Genome Sequencing Consortium (IWGSC) [4]. In the Fasta header of these models, information about chromosome number, genome and the long and short arm of the chromosome is present. I separated all 99386 gene models on the basis of different A, B and D genomes to which they belong with a locally written Python script. I found among 99386 models 32081 were from A, 34226 were from B and 33079 were from D genome. After this we made three different gene model databases of A, B and D genomes.1.2 million EST sequences of wheat were downloaded from dbEST [8] and were assembled using CAP3 [1] and Trinity [2] program as default parameters forming 32388 contigs [Supplementary file 7] and 522651 singlets by CAP3 and 46419 assembled sequences by Trinity respectively. We performed a BLASTn [5] search of 32388 contig sequences of CAP3 and 46419 assembled sequences from Trinity against three different gene model databases of A, B and D genomes and I extracted gene model id, evalue and percentage identity for every consensus sequence by locally written python script.This BLASTn was performed to get the information which is lacking about these sequences and was the "address in the genome" which is, among A, B or D which genome, which chromosome number and also where on the long or short arm of the chromosome on which these sequences are located on the basis of sequence similarity. From this we extracted genome gene model id, e-value and percentage identity for each consensus sequence by the same python tool.Genome location:I report 4 different 13 columns Microsoft excel tables for the location of our 4 different datasets [Supplementary file 1, Supplementary file 2, Supplementary file 3, Supplementary file 4, Supplementary file 5]. In the first it is from 1 to 32388 contig sequences assembled by CAP3 [1] program and in forth it is from 1 to 46419 sequences assembly by the Trinity [2] assembly program. The remaining columns are e-value, percentage identity, a query coverage percentage per highly similar pair of A genome gene model id, B genome gene model id and D genome gene model id. These 4 different tables show similarity with a gene model chromosome number and long/short arm information. We introduce a filter of equal or higher 90% percentage identity and query coverage percentage per highly similar pair in all 4 different data sets and we found that only 1.28% (416 out of 32388) of the contigs assembled by CAP3 [1], (1039 out of 46419) of the contig sequences assembled by the Trinity [2] were qualified [Figure 2]. For these sequences with at least 90% percentage identity and query coverage percentage per highly similar pair we are proposing the location of these sequences with chromosome number, genome and long and short arm. The results are in table number Supplementary file 5.
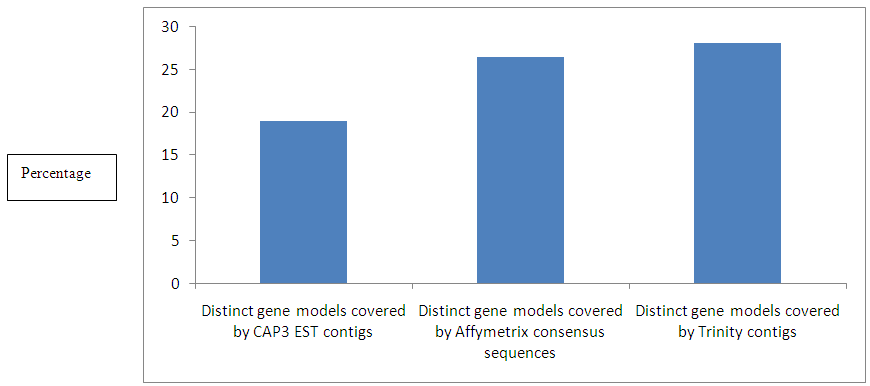 | Figure 1. Percentage of gene models covered by CAP3 EST contigs, Affymetrix consensus sequences and by Trinity contigs |
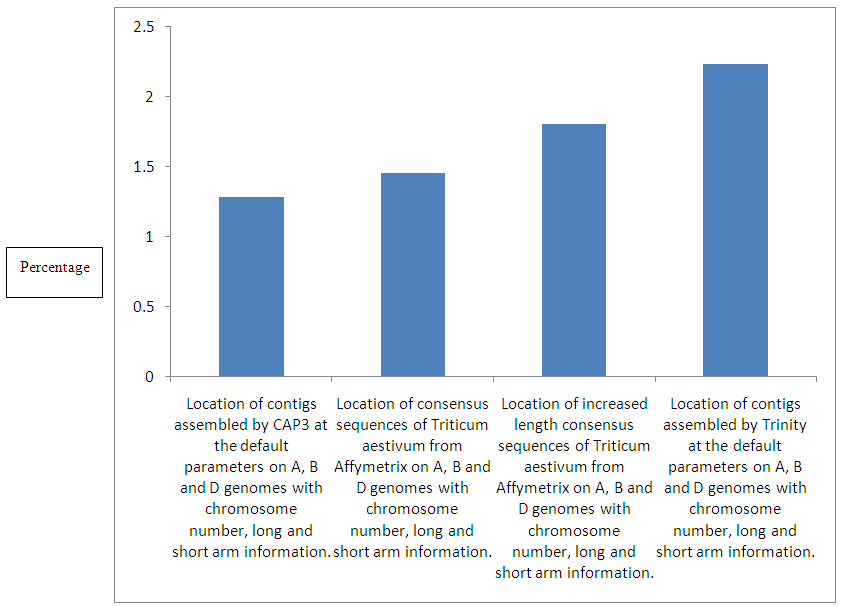 | Figure 2. Percentage of contigs assembled by CAP3 located on gene models of, Affymetrix consensus sequences, increased length Affymetrix consensus sequences and contigs assembled by Trinity |
 | Figure 3. Sequence alignment showing harboring of Affymetrix consensus sequence in increased length Affymetrix consensus sequences |
3. Discussion
- Percentage coverage of gene models and assembly:I performed a BLASTn [4] search of 32388 contigs assembled by CAP3 and 46419 assembled sequences by Trinity respectively against 99386 gene model sequence database. For 32388 contig sequences coming after assembling all EST sequences assembled by CAP3 [1] only 18.95% gene models [6] were covered and for the assembled sequences using Trinity [2] assembly program 28% gene models [6] were covered [Figure 1].
4. Conclusions
- Trinity [2] was found in the better then CAP3 [1] program because 0.95% more than the contigs assembled by CAP3 [1] were qualifying at least 90% threshold of percentage identity and query coverage percentage per highly similar pair.
ACKNOWLEDGEMENTS
- SS is thankful to the National Agri-Food Biotechnology Institute (NABI), Mohali, India.Data Archiving Statement: The data which is present in this paper is not submitted to any database.
Supplementary Material
- Supplementary file 1: This table shows similarity between 32388 contigs assembled by CAP3 program, gene models, chromosome number and location on the long / short arm. The first column is from 1 to 32388 contig sequences assembled by CAP3 [1] program ids remaining columns are e-value, percentage identity, a query coverage percentage per highly similar pair of A genome gene model id, B genome gene model id and D genome gene model id.Supplementary file 2: This table shows similarity between 46419 sequences assembled by the Trinity [2] assembly program, gene models, chromosome number and location on the long / short arm. The first column is from 1 to 46419 sequences assembled by the Trinity [2] assembly program remaining columns are e-value, percentage identity, a query coverage percentage per highly similar pair of A genome gene model id, B genome gene model id and D genome gene model id.Supplementary file 3: Selected sequence ids with gene model id, evalue, percentage identity, a query coverage percentage per highly similar pair having 90% percentage identity and query coverage percentage per highly similar pair of all 4 different data sets.Supplementary file 4: 32388 contig sequences assembled by the CAP3 program at the default parameters in FASTA format.Supplementary file 5: 46419 sequences assembled by the Trinity [2] assembly program at the default parameters in FASTA format.Appendix 1
Appendix 1.xls(13.7MB)Appendix 2
Appendix 2.xls(1.7MB)Appendix 3
Appendix 3.xls(7.0MB)Appendix 4
Appendix 4.xls(6.0MB)