-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2015; 5(1): 9-15
doi:10.5923/j.bioinformatics.20150501.02
Residue Frequencies and Conserved Phylogenetic Signatures in Amino Acid Sequences of Plant Glutathione Peroxidases, Indicates Habitat Specific Adaptation and Dictates Interactions with Key Ligands
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSayak Ganguli1, Abhijit Datta2
1DBT Centre for Bioinformatics, Presidency University, Kolkata, India
2Department of Botany, Jhargram Raj College, West Bengal, India
Correspondence to: Sayak Ganguli, DBT Centre for Bioinformatics, Presidency University, Kolkata, India.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Glutathione peroxidases in plants present one of the most interesting evolutionary enigmas due to the fact that aquatic members encode glutathione peroxidases with selenocysteine while land plants favour the amino acid cysteine. This analysis was performed to identify residue frequencies and conserved motifs in plant glutathione peroxidase sequences with the objective to identify possible evolutionary insights into this interesting group of stress enzymes. Phylogenetic footprinting was combined with parsimony analyses to identify true evolutionary signatures of the group. Following the identification of conserved phylogenetic signatures two essential ligands of the enzyme class-glutathione and hydrogen peroxide was used to study the interaction of the enzymes and the role of the conserved stretches in the interactions. Results indicate that these conserved stretches are important for maintenance of the structural and functional stability of these enzymes as well as interaction with key ligands.
Keywords: PlantGlutathione Peroxidase (GpX), Selenocysteine, Phylogenetic Footprinting, Evolutionary Signatures
Cite this paper: Sayak Ganguli, Abhijit Datta, Residue Frequencies and Conserved Phylogenetic Signatures in Amino Acid Sequences of Plant Glutathione Peroxidases, Indicates Habitat Specific Adaptation and Dictates Interactions with Key Ligands, American Journal of Bioinformatics Research, Vol. 5 No. 1, 2015, pp. 9-15. doi: 10.5923/j.bioinformatics.20150501.02.
Article Outline
1. Introduction
- Plants suffer from various kinds of oxidative stress as they are always under the influence of environmental factors [1, 7]. These factors contribute towards aerobic reactions which result in free radicals which serve as deleterious molecules in the cytoplasm. To counter these toxic compounds, plants have devised enzymatic defense mechanisms in the form of free radical scavenging enzymes such as superoxide dismutases, catalases, ascorbate peroxidases and Glutathione Peroxidases (GPx). Glutathione peroxidase (EC 1.11.1.9 and EC 1.11.1.12) family of enzymes are involved in catalyzing of the process of reduction of H2O2 or organic hydroperoxides to water or their alcoholic forms where reduced Glutathione (GSH) acts as an electron donor [10, 19]. At the molecular level, plant GPx genes are closely related to animal PHGPx and their corresponding proteins. Three widely conserved Cysteine residues, which are assumed to be essential for the enzymatic catalysis [17]. The most significant difference between mammalian and land plant glutathione peroxidases is the absence of selenosysteine in the catalytic centre of the later group [2, 3, 5, 6]. Moreover, the plant GPx gene family may comprise up to six members that are distributed in different subcellular compartments. Evolutionary analyses of plant GpX members have shown that their evolution was probably as a resultant of gene duplication and horizontal gene transfer events[16].
2. Materials and Method
2.1. Data Mining and Curation
- All glutathione peroxidase sequences which were available were downloaded from the NCBI Genpept resource and were carefully curated. Only those sequences which had more than 100 amino acids were selected for the study since protein motifs tend to occur in clusters and thus a minimum sequence length was required. Following this they were analyzed for their conserved domains to check whether all the sequences corresponded to the plant GpX family. No sequences were rejected at this step. Thus the final set of sequences numbered to 440 and the entire analyses was performed using them. It is important to mention at this point that distant homologues were not considered for the analyses.
2.2. Identification of Conserved Stretches
- The PERL (Practical Extraction and Reporting Language) script that was formulated produced a total of 175 positions from the multiple sequence alignment that was provided to it. This MSA was generated using Phylip and then validated using SEAVIEW. Once the positions were generated they were matched with the consensus sequence pattern that was obtained from the UGENE tool.
2.3. Phylogenetic Footprinting
- Phylogenetic footprinting [4] has been one of the most trusted methods to identify transcription factor binding sites in the upstream region of various genes since it was first used by Tagle et al. (1988) [24]. However, the authors decided to extend the repertoire of phylogenetic footprinting to amino acid sequences by combining maximum parsimony based tree generation methods along with basic multiple sequence alignment [9]. The basic steps of generating the footprint were as follows: ● Create a cluster of homologous sequences by applying homology search tools such as BLAST [18].● Since the cluster was already prepared a basic multiple sequence alignment [23] was performed using Phylip and was validated using SEAVIEW program. The multiple sequence alignment was constructed using Kimura distances and maximum parsimony was used to generate the cladogram [25]● A PERL program was designed which accepted the sequences in aligned format and calculated the residue frequencies for each position of the alignment. A host of frequency hits were retrieved: >20-90%● From the multiple sequence alignment a consensus sequence was generated using the UGENE [22] tool and the conserved regions represented in the alignment were analyzed
2.4. Molecular Dynamic Simulations and Docking
- Molecular dynamic simulations were performed with CABS FLEX server and docking was done using FLEX-X using an aquatic and a land plant glutathione peroxidase as the receptor and hydrogen peroxide and glutathione as ligand. Flexible docking was performed after specifying the grid for interaction at the catalytic centre for both enzyme structures. The following diagram summarizes the flowchart of the work (Fig. 1).
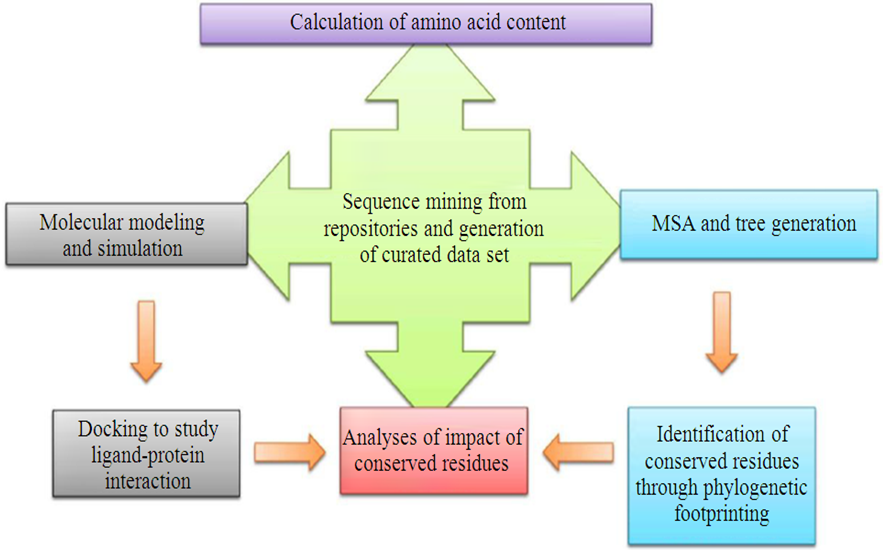 | Figure 1. Flow chart of Analyses |
3. Results and Discussion
3.1. Amino Acid Content
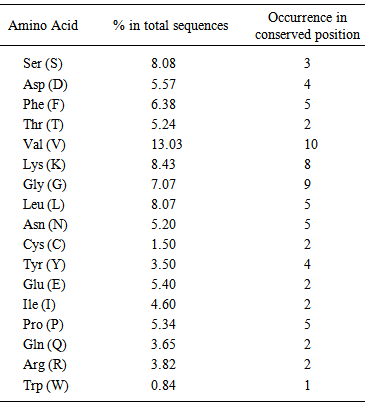
- Amino acid content and occurrence has been reported to be conserved from species to species. This conservation may be attributed to protein stability and expressivity of the proteome [11, 13, 14]. In case of individual protein families the occurrence and content govern the formation and maintenance of various motifs and domains. Recent studies have indicated that the mutational landscape of proteins can be correlated to the composition of amino acids. In this study Valine was found to be the most frequently occurring amino acid in the sequences under study. It was also observed that it was occurring in the conserved positions more often. The next most frequent amino acids were found to be lysine and Serine (Fig. 2 and Table 1).
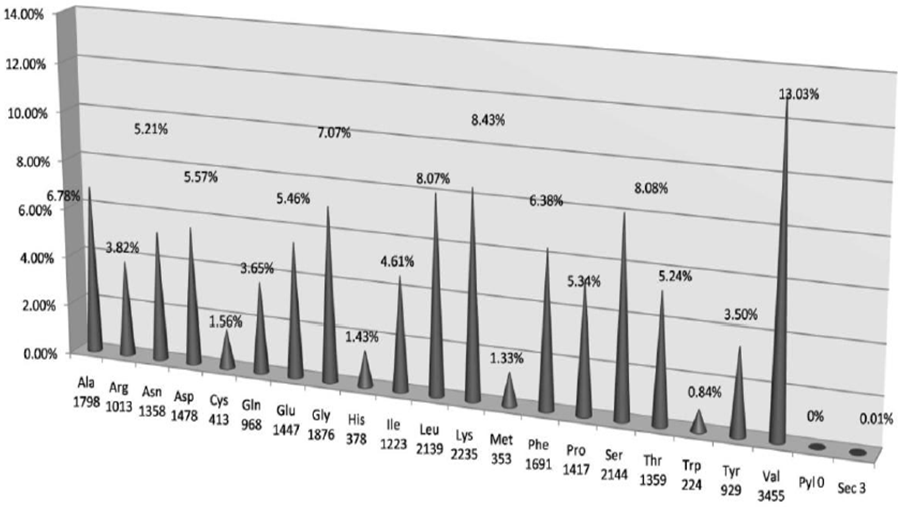 | Figure 2. Amino acid frequencies in conserved positions in glutathione peroxidase sequences |
|
3.2. Motif/Pattern Identification and Mapping of Conserved Positions
- It was found that most of the sequence positions present in UGENE were present in the positions that were obtained using the in house program (Fig 3 and 5). However, only those residues which had >60% frequency in their respective positions were found to be common in both the result sets. The gaps that were present between the residues (>60% frequency) were replaced with X (symbol for any amino acid) and a minimum length of 6 amino acids were selected as the window size. Now using this window size a sliding window approach was initiated which resulted in the generation of 3420 motif patterns. Now each of these patterns were searched against Prosite and eight conserved consensus patterns were identified which serve as phylogenetic signatures for plant glutathione peroxidases (Table 2). LYXKYK and EILAFPCNQFG patterns were the most dominant patterns with the largest number of hits (Fig. 3).
|
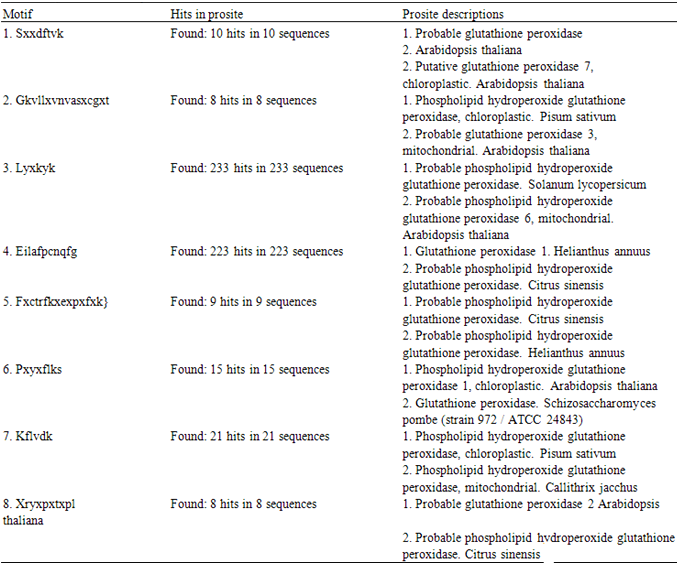
 | Figure 3. A portion of the consensus sequence generated indicating the sequence positions which correspond to phylogenetic signatures |
3.3. Phylogenetic Analyses and Comment on Evolution of Plant Glutathione Peroxidases
- The phylogenetic analysis reveals that GPXs in plants exhibit two main groups (Fig. 4)-one group which belongs to the aquatic members while the second group belongs to terrestrial land plants. A further interesting fact is two Cysteine residues were found to occur in the EILAFPCNQFG and the FXCTRFK motifs which have been reported to be part of the interacting residues of this enzyme group. We have earlier reported that these cysteine residues are important moieties which hydrogen bond with the respective ligands of glutathione peroxidases [12]. This divide is justified as several reports and analysis of amino acids sequences reveal that GPXs of aquatic members contain selenocysteine while those of land plants do not [8, 15]. Selenoproteins are absent in higher plants but green algae Chlamydomonas sp and Ostreococcus sp. possesses 12 and 26-29 selenoproteins respectively [20]. The lack of selenoproteins in land plants is a well documented phenomenon [15, 21]. The most accepted explanation states the conversion of anaerobic earth to aerobic earth through oxygenation and the selection of a more stable and less easily oxidizable forms of glutathione peroxidases. This analyses further strengthens the view with the observations that the identified phylogenetic signatures are important protein landmarks and their presence ubiquitously in case of both land and aquatic members indicate that possibly the evolution of glutathione peroxidases could have occurred along a modular assembly line.
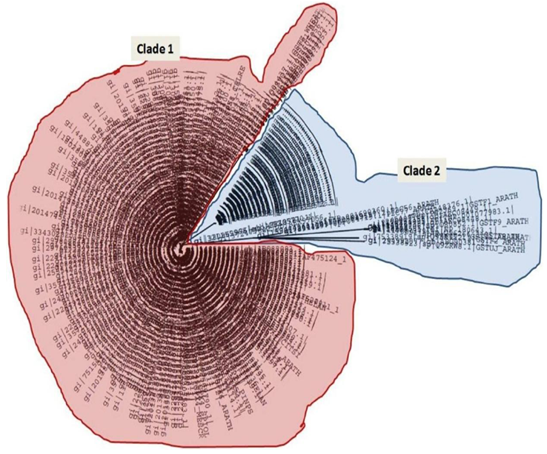 | Figure 4. Phylogenetic tree showing two distinct clades (Clade 1-Land plant GpXs and Clade 2-Aquatic GpX members) |
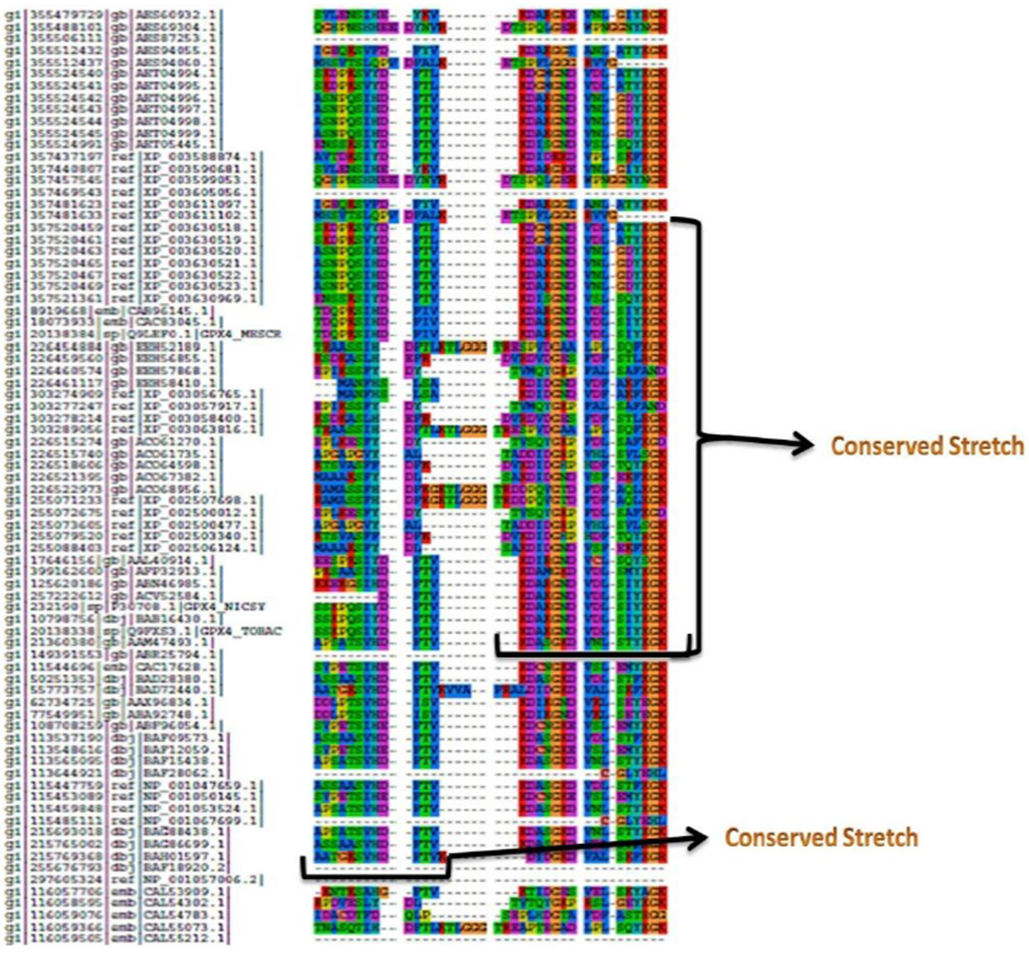 | Figure 5. A portion of the alignment of glutathione peroxidase sequences with the highest levels of sequence conservation (possible phylogenetic signatures) |
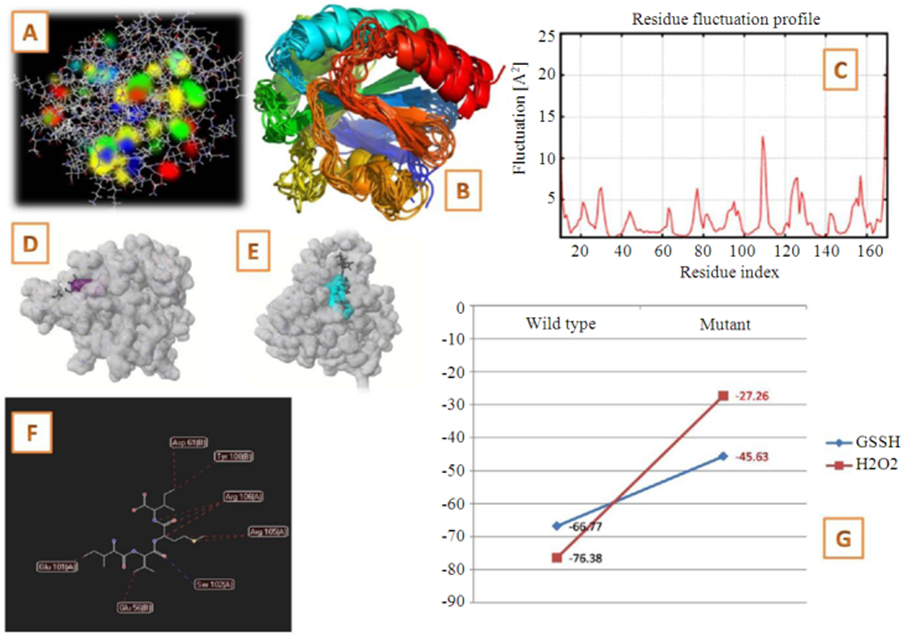 | Figure 6. Results of Structural and Interaction studies. A: Molecular model of glutathione peroxidase with its electron dense residues. B: Simulated structural alignment; C: Residue fluctuation profiles of the protein; D and E: Interactions of glutathione and hydrogen peroxide with the land plant and aquatic members respectively; F: Interacting residues following mutation; G: Changes in free energy of interaction of glutathione peroxidase with glutathione and hydrogen peroxide |
4. Conclusions
- Our analyses strengthens this hypotheses as the conserved residues identified in land plant lineages possess cysteine at the catalytic residue cluster, while in case of aquatic members it is replaced by selenocysteine. Both these residues are part of the conserved residues that were identified in the respective lineages thus showing that transition of selenocysteine to cysteine as a necessary adaptation for land plant transition. The involvement of the conserved residues in interactions also establishes their importance as functional phylogenetic signatures.
ACKNOWLEDGEMENTS
- The authors acknowledge the Department of Biotechnology, Ministry of Science and Technology for the funds provided to maintain the facilities at the DBT - Centre for Bioinformatics, Presidency University, under the BIF – BTBI scheme.