-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2015; 5(1): 1-8
doi:10.5923/j.bioinformatics.20150501.01
Theoretical Dynamics and Energetics of HLA-A2/SLYNTVATL Interaction
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOmotuyi I. Olaposi1, 2, Hamada Tsuyoshi3
1Department of Pharmacology and Therapeutic Innovation, Nagasaki University, Nagasaki, Japan
2Department of Biochemistry, Adekunle Ajasin University, Akungba Akoko, Nigeria
3Nagasaki Advanced Computing Center, Nagasaki University, Nagasaki, Japan
Correspondence to: Hamada Tsuyoshi, Nagasaki Advanced Computing Center, Nagasaki University, Nagasaki, Japan.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Human leukocyte antigen (HLA) class 1 is functionally involved in the recognition, binding and presentation of nonapeptide epitopes derived from proteasome-digested viruses and bacteria to T-lymphocytes. This study focused on key structural dynamics and free energy cost of HLA-A2- binding to a nonapeptide epitope (SLYNTVATL) using a combined 450 ns full atom molecular dynamics simulation. Data analysis showed that SLYNTVATL is not stable as an α helix in solution but adopts turn conformation with main chain RMSD value less than 5.0 Å from the starting structure within the last 30 ns of simulation. Clustering of the conformations sampled during the simulation also revealed that only 0.8% population shared structural resemblance with the X-ray-resolved, HLA-A2-bound nonapeptide with C-α RMSD value of 0.638 Å. The data further support that SLYNTVATL binding within the HLA-A2 groove may be preceded by rapid desolvation, induced fit rearrangement of the HLA-A2 groove atoms to arrest the first three (SLY) nonapeptide residues, while structural complementarity may account for the contact with the last four (VATL) nonapeptide residues. -3.00 kcal /mol was estimated for HLA-A2-SLYNTVATL binding. The predominant biophysical change within the binding-groove of HLA-A2 following SLYNTVATL binding was an approximately 3.0 ~ 4.0 Å expansion of the HLA α-helix which allowed an equilateral latching of the peptide between the α-helix and the β-barrel sheets. Finally, in bound conformation, the correlated motion within the groove is arrested in β-2 microglobulin dependent manner.
Keywords: HLA-A2, Nonapeptide epitopes, Structural dynamics, Free energy, Correlated motion
Cite this paper: Omotuyi I. Olaposi, Hamada Tsuyoshi, Theoretical Dynamics and Energetics of HLA-A2/SLYNTVATL Interaction, American Journal of Bioinformatics Research, Vol. 5 No. 1, 2015, pp. 1-8. doi: 10.5923/j.bioinformatics.20150501.01.
Article Outline
1. Background
- Human major histocompatibility complex (MHC) is a functionally diverse human leukocyte antigen (HLA) system which orchestrates CD8+-mediated cytotoxicity (MHC class I-HLA-A, B and C), activates antibody generation through interaction with CD4+-T helper cells (MHC class II) or functions as immune-modulatory cytokines and key members of the complement system (HLA class III). MHC class-I and II require binding and presentation of peptide antigens (Tax peptide) for their functions, while class I presents nonapeptide epitopes derived from proteasome-digested viruses and bacteria, class II is known to bind epitopes of extracellular origin. Here, the focus will be on class I MHC [1]. Structurally, histocompatibility antigen (class I) expressed on human cell membranes is a heterodimer comprising α (α1, α2 and α3) [2] and β2-microglobulin [3]. The β2-microglobulin non-covalently interacts with membrane-proximal α3. At the membrane-distal end of α3, eight antiparallel β-strands serve as a bridge-like scaffold upon which α1- α2-helices are latched forming the peptide-binding groove [2]. Post bacterial or viral protein digestion and release into the cytosol by the proteasome complex [4], it is not known whether the Tax-peptides already adopt bound conformation within the cytosol, during endoplasmic reticulum transportation by transporter associated with antigen protein or within the endoplasmic reticulum before loading [5]. Within the last two decades, methods for HLA system manipulation geared towards peptide-based vaccine development strategy have been on a steady rise [6, 7] and its success is owed to the emerging methods for the characterization and classification of MHC alleles into supertypes [8] and the crystallization of some of the HLA/tax peptide complex [9-12]. Although, the understanding of the subtle differences underlying antigen presentation and T-cell specificity has been broadened due to increasing number of HLA-related 3D structures deposited at the protein data bank (PDB) however, very little dynamical details can be gleaned from these strictures thus, necessitating the use of molecular dynamics simulation approach for generating complementary information. For instance, the conformational changes associated with T-cell receptor recognition and binding of HLA-DR4-peptide complex had been studied [13]. Similarly, a related study provided theoretical evidence in support of the roles of α-3 and β-2 microglobulin in the binding of tumour-specific "GVYDGREHTV" peptide to HLA-A*0201 [13]. Here, using molecular dynamics simulation method, we studied whether HIV-derived nonapeptide epitope "SLYNTVATL" readily adopts the HLA-A*0201(HLA-A2)-bound state conformation in a free state, the structural dynamics associated with SLYNTVATL binding and the free energy of SLYNTVATL-HLA-A2 interaction using the weighted histogram analysis and Jarzynski’s approximations [26].
2. Methods
2.1. Starting Structures
- The atomic coordinates for (HLA)-A2-SLYNTVATL complex was retrieved from the PDB (Figure 1A, PDB ID: 2V2W) [15]. Prior to molecular dynamics (MD) simulation, (HLA)-A2-SLYNTVATL complex was inserted into phosphatidylcholine (POPC) membrane in order to mimic the overall physiological setup. To establish whether free SLYNTVATL nonapeptide can sufficiently sample bound conformation in solution, an initial full alpha-helix SLYNTVATL nonapeptide (figure 1B) was built using Molecular Operating Environment (MOE) [16] with an estimated 4.259 nm alpha carbon root mean square distance (RMSD) from the HLA-A2-bond conformation.
2.2. Molecular Dynamics (MD) Simulation Procedure
- Groningen Machine for Chemical Simulations (GROMACS) version 4.6.2 [17] was used to perform full-atom MD simulations using GROMOS-53a6 all atom force-field parameter set [18]. The amino and carboxyl termini of the protein and peptides were capped with NH3+ and CO2- groups and solvated in explicit SPC-water box [19] in neutralizing Na+ /CL- ions (0.15M). Each soup (free peptide or HLA-peptide complex) was initially minimized and equilibrated at constant number of particles (N), temperature (T) and pressure (P) with full position restraint for 100 ns. During NPT equilibration and production run, protein and non-protein atoms were coupled to their own temperature baths at 310 K using the Berendsen algorithm [20] and the Berendsen barostat was used to maintain the pressure at 1.0 bar. Energy of each soup was minimized using the steepest descents method at a time-steep of 2 fs using LINCS algorithm for bond constraint [21]. Particle-Mesh Ewald (PME) method was used for long-range electrostatic interaction of particles using a Cut-off of 10 Å (5 Å for peptide simulation) [22]. All calculations were performed on Nagasaki University DEGIMA supercomputer (DEstination for Gpu Intensive Machine clusters [23].
2.3. Data Acquisition Method
- For Clear understanding of the time for data acquisition in each of the system simulated here we present figure 1C. Following position-restrained NPT simulation for 100ns, the first 30 ns of the unrestrained simulation were considered as equilibration in both systems. For the peptide only system, data were acquired after additional 30 ns. For HLA-peptide system, we extracted a low free energy coordinate from the simulation after 60ns and removed the peptide coordinate while the remaining HLA coordinate was subjected to brief 20 ns NPT equilibration, 30 ns restrained MD equilibration followed by data acquisition for 40 ns. In total, peptide (100 ns, 30 ns and 30 ns), HLA-peptide (100 ns, 30 ns and 70 ns) and HLA-A2 (20 ns, 30ns and 40 ns) were simulated at a combined time of 450 ns.
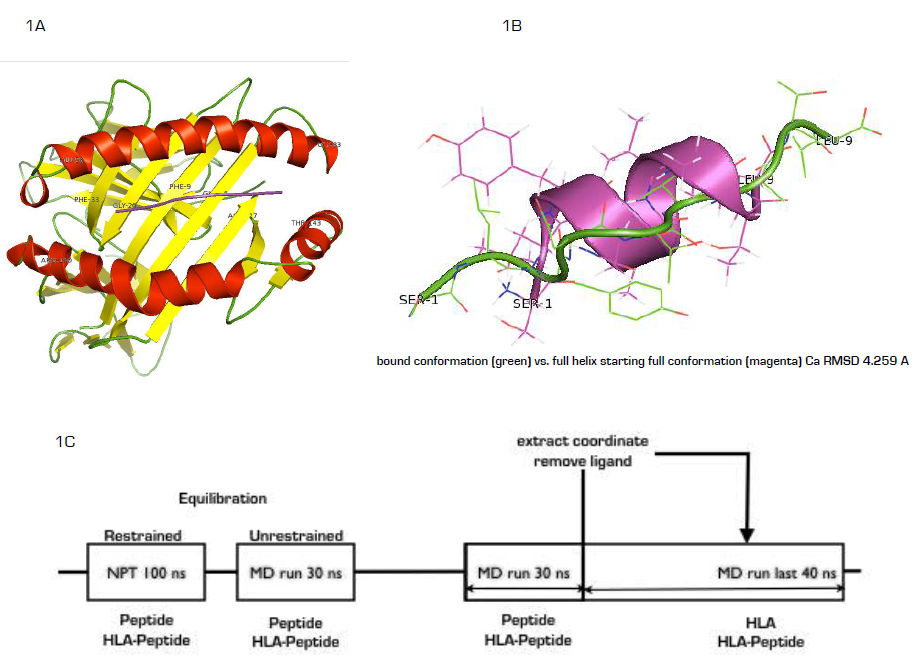 | Figure 1. (A) Ribbon representations of SLYNTVATL-HLA-A2 complex showing latched peptide within the binding groove. (B) The C-alpha RMSD of the representative of the highest structure population of the cluster (green, 98%) showing turn conformation at 4.259 Å from the starting helix conformation (purple) (C) The simulation steps from equilibration to unrestrained MD showing the time-lapse in nanosecond |
2.4. Data Analysis Tools
- Computation of the distance-based root-mean-square deviations (RMSD) of structure with time was done using the g_rms algorithm [17], the center-of-mass distance between particles and the radius of gyration were computed using the g_dist and g_gyrate algorithm [17] respectively. Hydrogen bond formed between a pair of donor and acceptor spaced at 0.35 nm and an angle of 30o was calculated using the g_hbond [17]. The root mean square fluctuation (RMSF) of atomic positions of the residues was computed using the g_rmsf algorithm [17]. RMSD-based structure clustering was done using the g_cluster algorithm based on gromos cut-off of 1.0 nm [24]. Secondary structure was assigned to each residue of ProTa during the simulation using the DSSP algorithm [25]. Estimated radius of gyration and hydrogen bonding interaction or RMSD to selected clusters (see result) was projected to 2D free energy surface plot using the Mathematica software.
2.5. Free Energy Estimation
- Potential of mean force (PMF) was used to reconstruct the energy barrier for HLA-A2-peptide interaction from 40 configurations spaced at 0.1 nm along the reaction coordinate generated following 500 ps pulling of peptide from the binding groove at pull rate of 0.01 nm ps−1 with a spring constant of 800pN. Each frame was sampled for 5ns with a harmonic restraint of 1000 pN applied on the ligand. For each run, initial 1 ns data were also regarded as equilibration while the final 4 ns of the trajectory were used for constructing the PMF curve using the weighted histogram analysis method and Jarzynski’s approximations [26].
3. Result and Discussion
3.1. In Solution, SLYNTVATL adopts Different Conformation from HLA-A2-bound State
- As a key step in molecular recognition and ligand binding, several peptides acting as ligand may adopt different conformations in free and bound states [27]. When different conformations are adopted by a ligand, it is commonly interpreted as a biophysical process, which traps the ligand within an energy landscape suitable for receptor/enzyme/partner recognition and binding, in other cases, initiating binding orchestrates conformational transition into a more energetically metastable state [28]. The simulation data showed that SLYNTVATL nonapeptide existed as two major conformational (99.2 and 0.8%,) population based on gromos clustering algorithm at 1.0 nm cut-off (figure 2A, B). Alignment of the representative structure of the 98% population with similar peptide co-crystalized with HLA-A2 (2V2W) showed 2.5 A RMSD while the average structure of the 0.8% population showed 0.639 A for C-α atoms; connoting that HLA-A2 bond SLYNTVATL conformation is scarce in free state. In order to further understand the energetic basis for the differential stabilities of the two conformations, we estimated the full atom RMSD of the entire population of peptide conformations generated during 30 ns production MD simulation to each of the average structures and their corresponding radius of gyration were projected into 3D energy landscape (2C&D). As expected, structures with full atom RMSD values between 0.45 and 0.60 nm are observably stable (purple, free energy = 0 kcal/mol) given that the radius of gyration is approximately 0.60 nm. In contrast, only dissimilar structures (RMSD > 0.60) from the population B are energetically stable enough to exist in solution during the simulation at the radius of gyration value of 0.60 nm. Starting with the HLA-A2-bound peptide conformation, the result showed that energetically stable HLA-A2 binding conformation could be attained when peptides in solution exist at full atom RMSD values between 0.35 and 0.42 nm and radius of gyration between 0.52 and 0.62 nm respectively. Such structures are not readily available in solution under our simulation conditions. Together, this data may provide a hypothetical proof that SLYNTVATL interaction with HLA-A2 may serve as the driving potential for the attainment of bond-conformation or the antigenic determinant conformation in the peptide. In more practical terms, T-cell receptor bias for only HLA-bound peptides rather than free antigenic peptides may be due to non-existing antigenic determinant conformation in free state [29, 30].
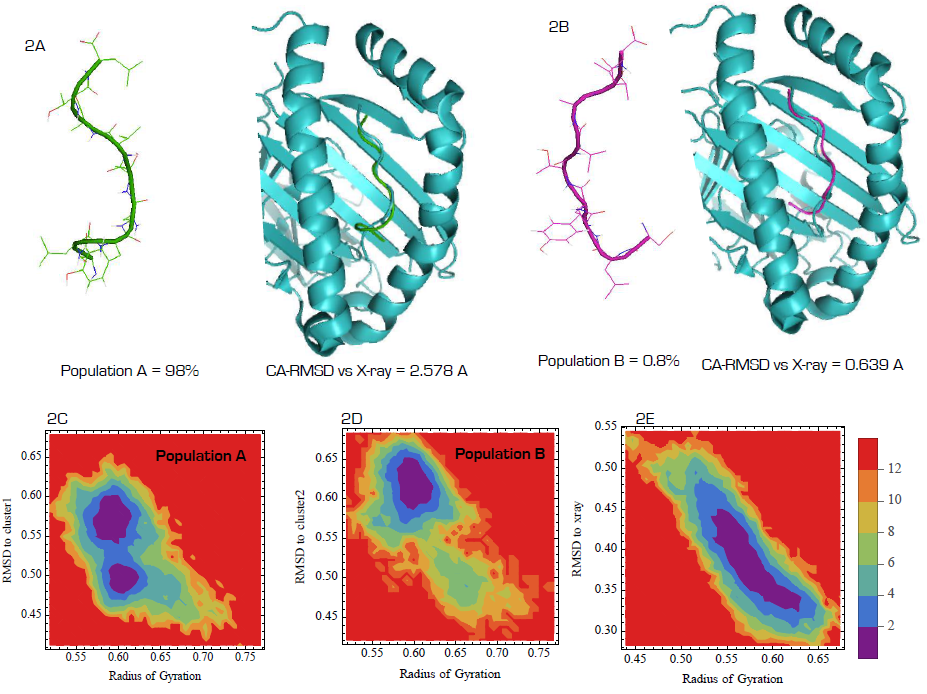 | Figure 2. Clustering analysis and energetics of cluster stability in solution. (A&B). In free state, 98% of SLYNTVATL existed at 2.578 Å RMSD from the bond structure while 0.8% existed at 0.639 Å. Energy landscape of total structures generated during simulation of free SLYNTVATL as transiting from the 2.578 Å RMSD structure (C), 0.639 Å structure (D) and the x-ray structure(E) |
3.2. Desolvation and Binding of SLYNTVATL Equilaterally within the Expanded HLA-A2 Groove
- To determine the biophysical processes associated with SLYNTVATL binding, first we estimated the number of water molecules resident at 0.35 nm radius of the free peptide in solution and compared the result with the amount found within similar radius in bound state. Interestingly, the free peptide appeared highly solvated with about 33 molecules of structural water. In bound state, less than 20 molecules of water were found at the same cut-off radius. This is in consonance with the principle of ligand desolvation prior to receptor interaction [31, 32]. Indeed, stability of highly polar molecules in water has been linked to thermodynamically favourable solvent-mediated intramolecular interactions [32] and overcoming the large desolvation penalties for HLA-A2 interaction may therefore depend on bond-breaking, bond-formation energy compensation, indeed, approximately 6 hydrogen bonds existed between HLA-A2 and the bound peptide (Fig. 3A) which serves to compensate the energetic changes. Since HLA-A2-SLYNTVATL complex is experimentally confirmed as stable [15], other factors such as entropy changes may have an overall stabilizing rather than destabilizing effect on the complex [33]. Next we determined the distance between the α-helices located within the peptide-binding groove [34]. The data revealed that in the absence of the tax peptide, the inter-helical distance is subtly maintained at 22 A, upon peptide binding, the space is expanded by approximately 3-4 A (Fig. 3B). Although this observation has not been experimentally documented, it may be interesting to note that mechanisms involved in TCR -MHC recognition has been traced to flexibility in both TCRs and peptides and more recently, mobility in the MHC groove in a peptide-dependent manner [35]. Hypothetically, such 3-4 Å anti-parallel shift (Fig 3C, cyan ribbon is the average HLA-A2 structure within the last 5 ns of the simulation without the ligand, while the green is similar structure bound to SLYNTVATL) may be required for geometrical alignment and contact optimization during TCR interaction [36]. Lastly, we determined whether the peptide showed skewered positioning towards any of the two helices or the eight antiparallel β-strands bridging the groove [34]. The data showed that throughout 70 ns of simulation, the peptide showed a unique equilateral localization (figure 3D). Hypothetically, TCR recognition of HLA-peptide complex may depend strictly on these subtle geometries. In fact, peptide must be superficially displayed within the groove for TCR recognition [36]. The data here can be summed up as shown in figure 3E as peptide desolvation, HLA-binding, groove helix displacement and equidistantly located within the groove.
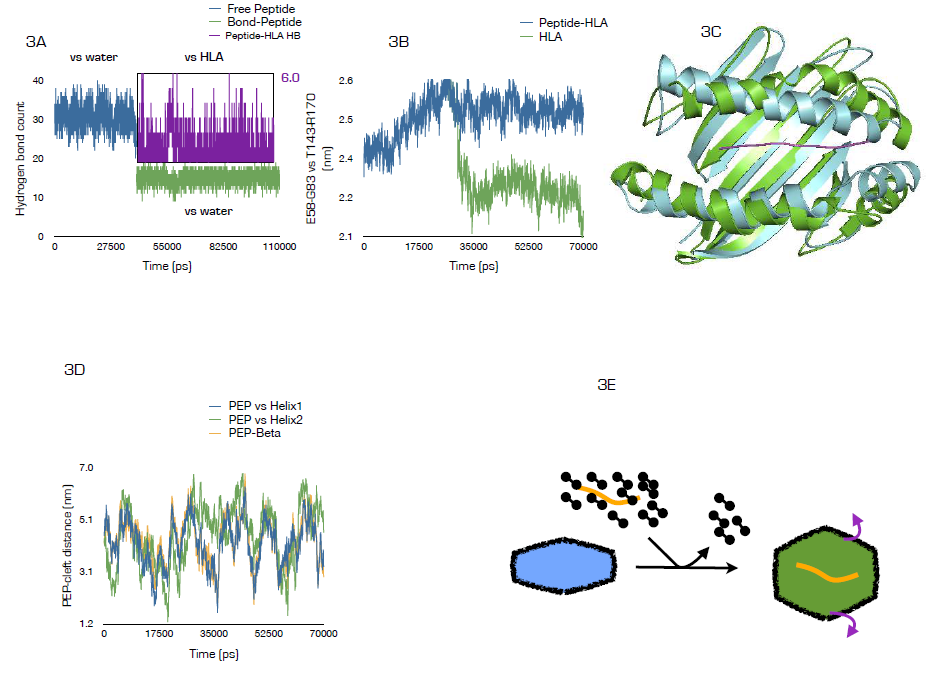 | Figure 3. Mechanism of Tax-peptide HLA interaction. (A) Number of water molecules found within 3.5 Å radius of the peptide in free (blue) and bound (green) states and the hydrogen bound number between HLA and SLYNTVATL within the last 70 ns of simulation. (B) Inter- alpha helix1-helix2 distance in the presence or absence of Tax peptide showing ~ 30 A increase in a1-a2 distance following tax-peptide binding. (C) Superimposition of the average HLA-A2 structure within the last 5 ns of the simulation (cyan) without the ligand, while the green is similar structure of HLA bound to SLYNTVATL taken at the same time frame. (D) Tax-peptide is equidistantly located within the groove during simulation as indicated by the change in center of mass distance between the peptide and α1, α2, and the lining residues of the eight antiparallel β-strands. (E) Mechanistically, binding of the Tax peptide within the groove is associated with desolvation, expansion of the groove and equidistant localization of peptide within the groove |
3.3. SLYNTVATL Perturbs the Dynamics of HLA-A2 without Altering the Residue Secondary Structures
- Molecular dynamics simulation allows the explorative comparison of the intramolecular dynamics of receptor proteins in the presence or absence of ligand though the cross-covariance analysis or dynamic cross-correlation matrix (DCCM) analysis [37]. Here, using the cross-covariance analysis (figure 4A & 4B), we showed that the HLA-A2 peptide-binding groove (red ribbon) showed anti-correlated motions (red), which is either arrested (white) or resolved as correlated motions (blue) in the presence of the peptide. Interestingly, the groove residues showed strong correlated motions relate to the non-covalently attached β-2 microglobulin (yellow ribbon) in peptide-free state. In the presence of the peptide, such motions were predominantly arrested (white). Although the dynamical function of the arrested groove-β-2 microglobulin may not be certain from the simulations, the data however agrees with previously reported β-2 microglobulin function as the major stabilizer of the groove [38]. Indeed, in the absence of β-2 microglobulin, the groove looses its peptide binding and retention functions [38, 39]. With the large arrest in local motions around the groove region, we expected a subtle change in the secondary structures of the residues. The data however does not support this hypothesis as no significant change in the secondary structures was observed in peptide free and bound HLA-A2 protein (fig. 4C). We then determined how HLA-A2 binding altered the psi and phi backbone conformational angles for each residue in the peptide (4D) using the Ramachandran plot. In free state, Ser-1 and Lue-2 existed as α helices while in bound state while they exist predominantly as β-sheet in free state. Tyr-3 in bound state is trapped as a β-sheet residue while it exists equilibrium α-helix/β-sheet states in free state. Asn-4 has no distinctive conformational angle bias in both free and bound state but exists in α-helix/β-sheet equilibrium. Tyr-5 showed a unique eclipse conformation in bound state; while it exists in dynamic α-helix/β- sheet equilibrium in free state. Val-6 and Leu-9 are biased for β-sheet conformation regardless of their interaction status. Interaction with HLA-A2 causes Ala-7 to be trapped in an energetically unfavorable conformation from a dynamic α-helix/b-sheet conformation in free state. Tyr-8 existed in preferred α-helix in bound state but in free solution, it is a β-sheet. The data here suggest that 1-SLY-3 contact with HLA-A2 via induced fit whereby interaction forces the residues to abandon their initial conformational path to a new path dictated by HLA-A2. In contrast, 6-VATL-9 may interact by lock-and-key hypothesis as the required conformation for binding is already assumed prior to HLA-A2 interaction. Coincidentally, some experimental data are providing pointers to the conformational dynamics of peptide cum HLA binding [15, 40-41].
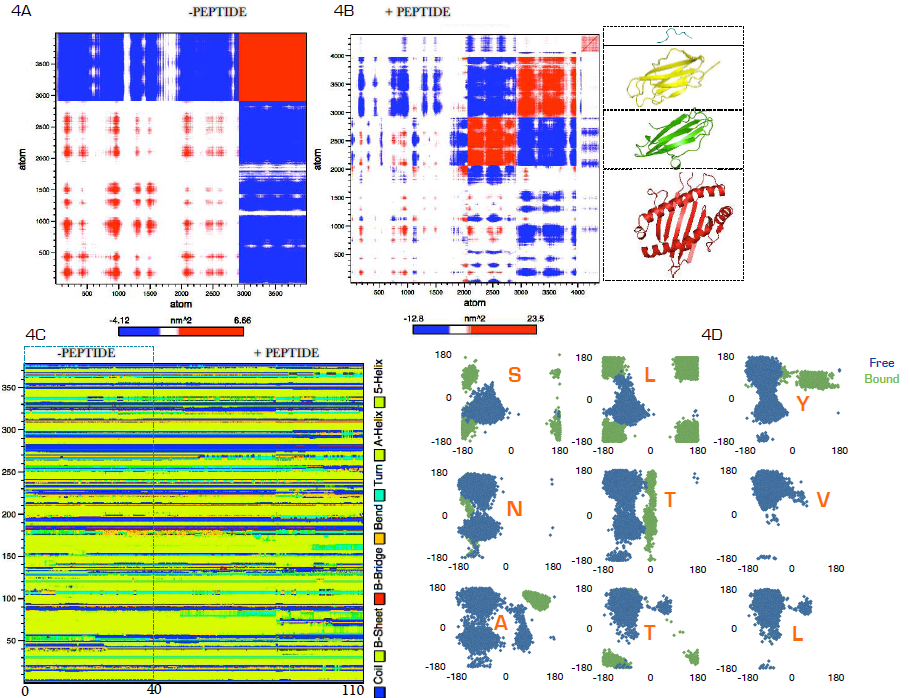 | Figure 4. Large-scale correlated motions, change in secondary structure and stability of HLA-A2 upon Tax-peptide interaction.(4A&B) Biding of Tax-peptide causes large inhibition of anti-correlated atomic motions in HLA-A2 molecule. (C) The inhibition of anti-correlated atomic motions in HLA-A2 molecule is not associated with bulk change in the secondary structure evolution of the residues. (D) Tax-peptide dihedral transition analysis |
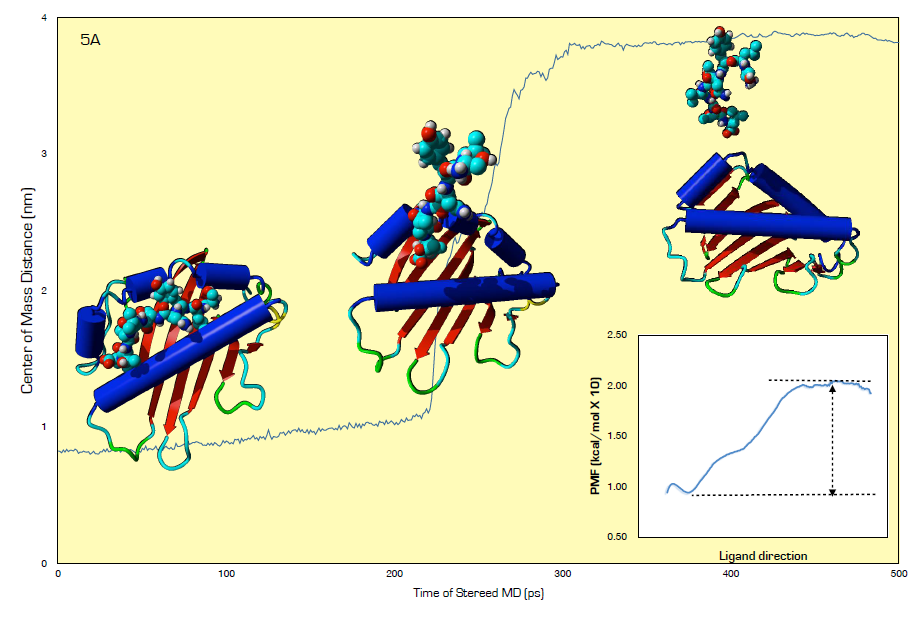 | Figure 5. Steered molecular dynamics analysis and weighted histogram analysis method for PMF calculation for HLA and SLYNTVATL free energy of binding estimation |
3.4. Energetics of SLYNTVATL/HLA-A2 Interaction
- The earliest strength of interaction between HLA-A2 and Tax peptide was conducted using thermal shift assay [40] and more recently, surface plasmon resonance has been used to analyze the interaction between G10 TCR and HLA-A2–SLFNTVATL or HLA-A2–SLYNTVATL [15]. Here, using steered molecular dynamics (SMD)(Fig. 4A) and umbrella sampling simulations of 40 different configurations spaced at 0.1 nm diameter, we extracted the potential of mean force (PMF) using the Weighted Histogram Analysis Method (WHAM) [26]. The difference between the minima and maxima plateau of the PMF curve approximates the Gibbs free energy (ΔG) for the binding of HLA and SLYNTVATL (Fig. 4B). The data showed that only -3.00 kcal/mol energy barrier prevents SLYNTVATL from binding to the peptide-binding groove of HLA-A2. Comparatively, if by using the surface plasmon resonance technique, a value of -7.7 kcal/mol activation energy was required for G10 TCR/HLA-A2–SLYNTVATL interaction [15], it would seem that for a lesser complex but readily interacting partners such as SLYNTVATL and HLA-A2, therefore, -3.00kcal/mol energy would be hypothetically reasonable. Thus, providing evidence in support of low activation energy requirement for SLYNTVATL/HLA-A2 interaction.
4. Conclusions
- This study suggested that the tax peptide (SLYNTVATL) investigated adopted different conformation in HLA-A2-bound and free states such that antigenic determinant conformation in free state is scarce. This observation has two broad clinical implications. First, in terms of autoimmune disorders, this serves as conformational sieve, which allows HLA-A2 to adequately distinguish self and non-self peptides before the initiation of CD8+-mediated cytotoxicity. Secondly, it provides a basis for the failure of many synthetic tax peptides to elicit immune response as the conformation for binding may have been adopted during digestion stage. Another key finding in this study was the binding mode of the tax-peptide within HLA-A2. Extensive desolation of the HLA-A2 groove, a slight superficial displacement of peptide, and a unique equilateral adjustment of the tax-peptide between the α-helix and the β-barrel sheets within the groove may force groove α helix expansion while arresting global correlated motion in β-2 macroglobulin dependent manner. These features may represent key geometrical properties for distinguishing tax peptide-bound HLA-A2 from the circulating free forms. Finally, our data also suggested that binding might proceed via induced fit at the first three residues and lock and key at the last four of the peptide with activation energy of less than -10.0 kcal.