-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2014; 4(1): 1-5
doi:10.5923/j.bioinformatics.20140401.01
Localized Smith-Waterman Algorithm for Fast and Complete Search of siRNA off-Target Sequences
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML1Department of Mathematical Science, University of Saint Joseph, West Hartford, 06117, USA
2Department of Chemical Biology and Center for Cancer Prevention Research, The State University of New Jersey, Rutgers, 08854, USA
Correspondence to: Hong Zhou, Department of Mathematical Science, University of Saint Joseph, West Hartford, 06117, USA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The Basic Local Alignment Search Tool (BLAST) is the most powerful and popular sequence alignment tool used in biological research, which includes the siRNA or shRNA design. Unfortunately, BLAST can overlook some significant homologies that may cause off-target effect for siRNA gene silence. Therefore, the Smith-Waterman alignment algorithm has been called upon, since it guarantees to find all possible mismatch alignments that may cause off-target effect. However, the Smith-Waterman alignment algorithm suffers from its inefficiency in searching through a large sequence database. This paper presents a two-phase homology search strategy that preserves the strength of Smith-Waterman alignment algorithm while shortening its running time. In the first phase of this algorithm, selected siRNA sequences are divided into multiple mutually disjointed substrings and each is used to scan the sequence database for perfect matches against other genes. Only the sequences that have a perfect match to substrings (of a given siRNA) are kept for the second phase. The second phase is the bona fide Smith-Waterman procedure. During this phase, the algorithm only checks the local vicinity sequences where a substring lands on a perfect match. This two-phased arrangement of the algorithm significantly improves the efficiency of the original Smith-Waterman algorithm by concentrating the search on localized regions instead of the whole genome database.
Keywords: siRNA, shRNA, Smith-Waterman, Sequence Alignment, Off-Target
Cite this paper: Hong Zhou, Hong Wang, Localized Smith-Waterman Algorithm for Fast and Complete Search of siRNA off-Target Sequences, American Journal of Bioinformatics Research, Vol. 4 No. 1, 2014, pp. 1-5. doi: 10.5923/j.bioinformatics.20140401.01.
Article Outline
1. Introduction
- RNA interference (RNAi) has become a powerful technique to knock out/down the expression of target genes for gene function studies in various organisms[3, 5, 16]. To employ this technique, the very first step is to design target-specific small interference RNA (siRNA) or small hairpin RNA (shRNA) that is homologous to the target mRNA. Because of the predictability of RNAi based on its matching target sequence[2, 5, 7, 9-11, 14, 15, 19, 22, 25, 26], quite a few studies have been devoted to computer-guided algorithms to design effective small interfering RNA (siRNA)[4, 6, 12, 15, 20, 25, 26]. However, a critical requirement in siRNA design is to guarantee that the designed siRNA sequences are free of off-target effect. Although the actual mechanism of off-target effect is still unknown, it has been demonstrated that a partial sequence homology between siRNA and its unintended targets is one of the contributing factors[8, 18, 21]. It has also been suggested that if an introduced siRNA has less than 3 mismatches with an unintended mRNA, it would likely knock down the expression of this mRNA in addition to its intended target which shares 100% homology with this siRNA sequence[11, 15]. Currently, some available siRNA design tools use BLAST to identify siRNA candidates that may cause off-target effect. BLAST, although very fast, is not the best algorithm designed for this type of task since it overlooks significant sequence homologies[15, 24, 27]. As an alternative, Smith-Waterman search algorithm has been employed together with BLAST by some design tools to identify all possible off-target sequences[15, 27]. Smith-Waterman algorithm[23] utilizes a dynamic programming approach to identify the local optimal alignment between two sequences. It guarantees to locate the existing optimal alignment based on a scoring system with a set of scores assigned to a match, a substitution, a deletion, and an insertion. Given two sequences with length of m and n, the computational complexity of Smith-Waterman algorithm is O (mn). Since the off-target search for siRNA sequences must be conducted completely through a given sequence database (which is usually large), the Smith-Waterman algorithm alone becomes very time-consuming and impractical for this task. Derived from dynamic programming, BLAST and FASTP improved the searching efficiency greatly by using a pre-constructed lookup table to find the locally similar regions between two sequences[1, 13, 17]. In BLAST, for example, given a sequence of m letters long, this sequence can have m−w+1 contiguous words each with w letters in length. These words can be scanned through the lookup table to find statistically significant word matches upon which a final alignment can be achieved. Though BLAST is very fast, the tradeoff for its efficiency is that it sometimes overlooks homologous sequences that can cause off-target effect[15, 24, 27]. In this paper, we present a two-phase homology search algorithm for siRNA off-target search that combines the effectiveness of BLAST concept and the thoroughness of Smith-Waterman algorithm. In this algorithm, we first divide the siRNA sequence into multiple mutually disjointed substrings based on the mismatch cut-off for an off-target homology (a mismatch is defined to be either a substitution, a deletion or an insertion hereafter). An off-target homology is defined to be an off-target sequence that has less than a predefined number of mismatches (mismatch cut-off) with the siRNA sequence. When and only when a substring of the siRNA sequence finds a perfect match, Smith-Waterman dynamic programming is used to examine the local vicinity region of the matched sequence. This algorithm does not construct any lookup table from the whole genome sequences, though it significantly improves the searching efficiency by guiding the most time-consuming core Smith-Waterman alignment on the local regions that need to be further examined. Yet unlike the BLAST algorithm, this algorithm does not miss any homologous sequences in siRNA that may cause off-target effect.
2. The Two-Phase Algorithm
- As a dynamic programming approach, Smith-Waterman algorithm finds the optimal alignment gradually by means of simple recurrences. To complete the alignment process between the two sequences of length m and n, a table of size
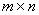 must be constructed. Thus, the computational complexity is of order O(mn). When searching for possible off-target alignment, a siRNA sequence must be aligned against all expressed sequences (mRNAs) except the target gene itself (or gene slice variants). In the human mRNA RefSeq database used for this study, there are 68822 non-redundant sequences with an average length of 3452bp. Therefore, the general computational cost for a siRNA sequence of 21nt would be at least
must be constructed. Thus, the computational complexity is of order O(mn). When searching for possible off-target alignment, a siRNA sequence must be aligned against all expressed sequences (mRNAs) except the target gene itself (or gene slice variants). In the human mRNA RefSeq database used for this study, there are 68822 non-redundant sequences with an average length of 3452bp. Therefore, the general computational cost for a siRNA sequence of 21nt would be at least 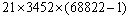 (suppose only aligning the sense strand of the siRNA sequence against other mRNA sequences). This cost-prohibitive computation makes it almost impossible to incorporate Smith-Waterman algorithm into routine siRNA design. To make Smith-Waterman algorithm practical in siRNA design, we propose a modified application of the algorithm, which was motivated by the following realization.For a siRNA sequence of length m, an off-target homology is defined as a sequence that has less than x mismatches when aligned against the siRNA sequence. Thus, after the siRNA sequence is divided into x equal substrings (as equal as possible), at least one substring must have a perfect match with the off-target region. For the remainder of this paper, let’s assume m=21 and x=3 unless stated otherwise. Under this condition, an off-target homology can only have a maximum of two mismatches, i.e., 0, 1, or 2 mismatches. When there are a maximum of two mismatches, no matter where the possible two mismatches are, at least one third of the siRNA sequence must have an exact match with the homological region as shown in figure 1.
(suppose only aligning the sense strand of the siRNA sequence against other mRNA sequences). This cost-prohibitive computation makes it almost impossible to incorporate Smith-Waterman algorithm into routine siRNA design. To make Smith-Waterman algorithm practical in siRNA design, we propose a modified application of the algorithm, which was motivated by the following realization.For a siRNA sequence of length m, an off-target homology is defined as a sequence that has less than x mismatches when aligned against the siRNA sequence. Thus, after the siRNA sequence is divided into x equal substrings (as equal as possible), at least one substring must have a perfect match with the off-target region. For the remainder of this paper, let’s assume m=21 and x=3 unless stated otherwise. Under this condition, an off-target homology can only have a maximum of two mismatches, i.e., 0, 1, or 2 mismatches. When there are a maximum of two mismatches, no matter where the possible two mismatches are, at least one third of the siRNA sequence must have an exact match with the homological region as shown in figure 1.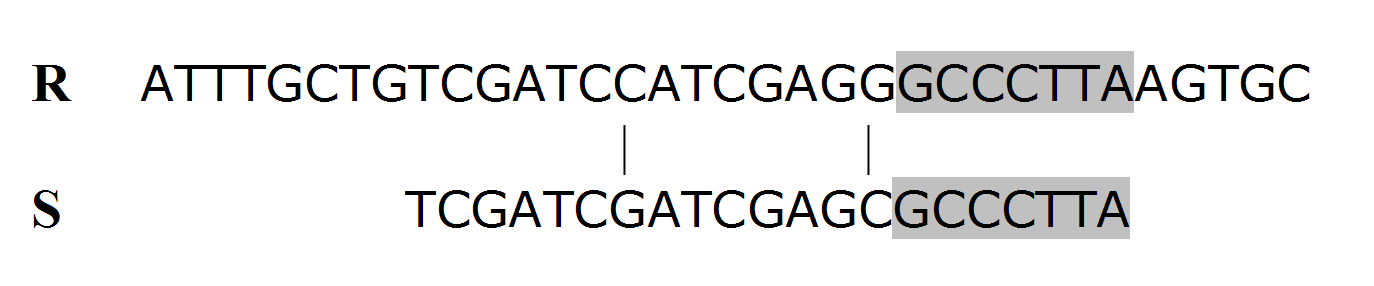 | Figure 1. When there are 2 mismatches between the siRNA sequence (S) and the off-target region (R), at least one of the three substrings of S has an exact match with R. The vertical bars mark the mismatches and the shaded substrings have the exact match |
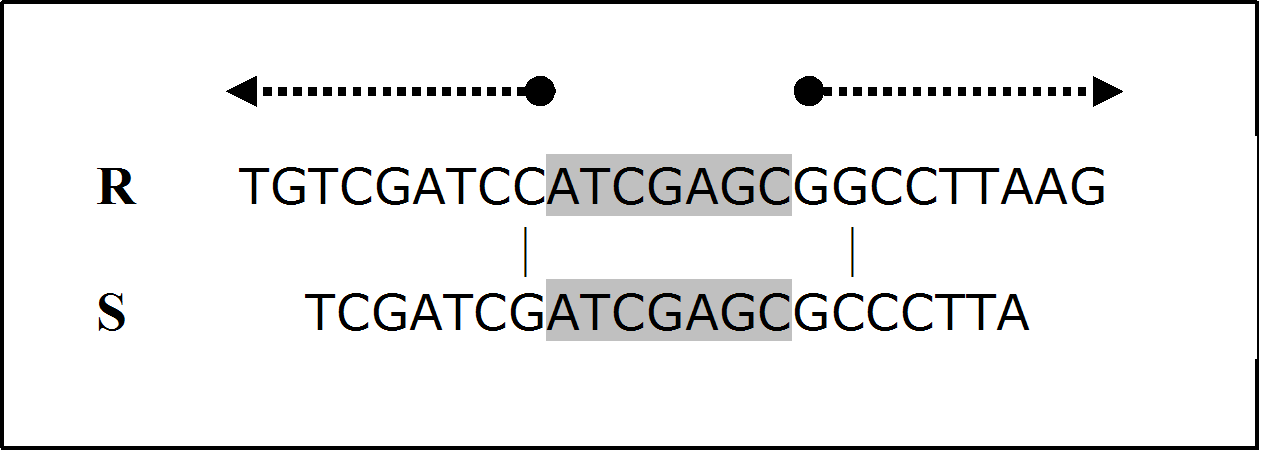 | Figure 2. When the substring in the middle has the exact match, the off-target region must be such a region that extends from the matched substring to both left and right enough base pairs to completely cover the siRNA sequence |
3. The Computational Complexity
- The total computational complexity of the proposed two-phase algorithm is the sum of the computational cost of the two phases. For the first phase, various string matching algorithms can be employed. As our implementation is in computer programming language Java, Java’s built-in string matching algorithm is used, which is a character by character matching procedure (brutal force). To search through a gene sequence, the maximum computational cost of this approach is of order
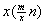 where m is the length of the siRNA sequence, n is the length of the searched gene sequence, x is the number of substrings, and
where m is the length of the siRNA sequence, n is the length of the searched gene sequence, x is the number of substrings, and 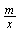 annotates the substring length. If we assume that each of the four different bases AGCT has an equal probability to appear at any given nucleotide position, then the probability for i consecutive bases to be matched is
annotates the substring length. If we assume that each of the four different bases AGCT has an equal probability to appear at any given nucleotide position, then the probability for i consecutive bases to be matched is 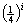 . However, in the character by character matching procedure, the second base is required to be compared only if the first base is matched, and the third base is required for comparison only if the first two bases are matched, and so on. Thus, the probability of comparing two bases of the substring is
. However, in the character by character matching procedure, the second base is required to be compared only if the first base is matched, and the third base is required for comparison only if the first two bases are matched, and so on. Thus, the probability of comparing two bases of the substring is 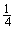 while the probability of comparing three bases of the substring is
while the probability of comparing three bases of the substring is 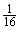 . Therefore the total computational cost for the first phase would be
. Therefore the total computational cost for the first phase would be | (1) |
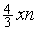 . The second phase is Smith-Waterman alignment algorithm conducted between the siRNA sequence and the potential off-target region whose maximum length is
. The second phase is Smith-Waterman alignment algorithm conducted between the siRNA sequence and the potential off-target region whose maximum length is 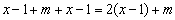 . The computational cost of this alignment procedure is therefore of order
. The computational cost of this alignment procedure is therefore of order 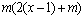 . However, the probability of finding a potential off-target region is only
. However, the probability of finding a potential off-target region is only 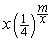 , so the actual computational cost of the second phase is
, so the actual computational cost of the second phase is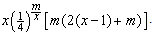 | (2) |
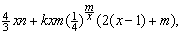 | (3) |
4. Results and Discussion
- The computational cost of Smith-Waterman alignment algorithm is linearly proportional to the length of siRNA sequence m. However, regarding the two-phase algorithm, as shown in equation (3), when x is fixed, larger m exponentially reduces the probability of phase two operations while it only linearly increases the computational cost of phase two operations, which can slightly improve the overall efficiency. As elucidated in figure 3 where x=3, the computational cost of Smith-Waterman alignment algorithm is linearly increased along with the siRNA length, while the computational cost of the two-phase algorithm is slightly decreased. Figure 3 also demonstrates the 50 – 100 times of efficiency gain of the two-phase algorithm.
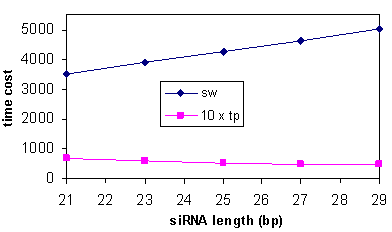 | Figure 3. The two-phase algorithm can gain 50 – 100 times in efficiency. sw: Smith-Waterman algorithm alone. tp: the proposed two-phase algorithm. The time cost value of the two-phase algorithm is multiplied 10 times for a better visual comparison |
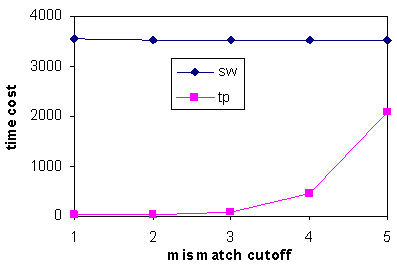 | Figure 4. The value of x impacts the efficiency of the two-phase algorithm. sw: Smith-Waterman algorithm alone. tp: the proposed two-phase algorithm |
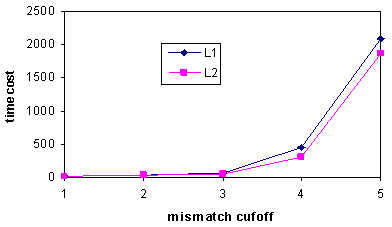 | Figure 5. Applying a sub-substring checking can further slightly improve the efficiency. L1: the two-phase algorithm; L2: the two-phase algorithm with sub-substring checking before phase two operation |
5. Conclusions
- The efficiency gain of the two-phase algorithm is achieved by setting up a first phase filter that relieves the burden for the most inefficient but reliable Smith-Waterman alignment algorithm. Though there are improved algorithms for the two procedures employed for the two phases in this study[29], the critical idea is to localize the Smith-Waterman alignment to regions where it is absolutely needed. Using siRNA design tool as an example here, we have demonstrated that the two-phase algorithm can achieve efficiency gain up to two orders of magnitude over the original Smith-Waterman alignment algorithm alone while retaining complete detection of off-target homology. Thus, the two-phase algorithm can be incorporated in the routine search for potential off-target regions in siRNA design, a task that BLAST algorithm cannot fulfill satisfactorily. In fact, this two-phase off-target searching algorithm played a critical role in making our computer-aided siRNA design tool capable for designing siRNA or shRNA on a whole-genome scale[27], helping verify that the shRNA designed by our tool can reach a success rate of over 70%[28].
ACKNOWLEDGMENTS
- The authors would like to thank Dr. Joseph Manthey for his valuable discussion about the computational cost analysis and his critical reading of this manuscript.