-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2013; 3(3): 62-71
doi:10.5923/j.bioinformatics.20130303.03
In Silico Prediction of Mechanism of Erysolin-induced Apoptosis in Human Breast Cancer Cell Lines
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFahad M. Al-Khodairy1, M. Kalim A. Khan2, M. Kunhi3, Manogaran S. Pulicat4, Salman Akhtar2, Jamal M. Arif5
1Carcinogenesis Section, Department of Molecular Oncology, KFSH & RC, Riyadh, 11211, KSA
2Department of Bioengineering, Integral University, Lucknow, 226026, India
3Cardiovascular Research Program, KFSH & RC, Riyadh, 11211, KSA
4Stem Cell & Tissue Re-engineering Program, KFSH & RC, Riyadh, 11211, KSA
5Department of Biochemistry, College of Medicine, University of Hail, Hail, KSA
Correspondence to: Jamal M. Arif, Department of Biochemistry, College of Medicine, University of Hail, Hail, KSA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Targeted therapy has been gaining momentum to be a promising strategy for drug development research. Natural compounds and their synthetic analogues have provided several promising anticancer drugs. Erysolin present in the Eruca sativa better known as rocket plant has been scarcely studied. In the present study, we assessed the anticancer potential and underlying mechanism(s) of action of erysolin in human breast cancer cell lines (MCF-7 and MDA-MB-231) using the in vitro and/or in silico experiments. Incubation of these cell lines with erysolin (10 and 50 µM) for 72 h resulted in 50-70% apoptosis in both cell lines at 50 µM with concomitant decrease in the cell viability. However, 10 µM erysolin induced 20% apoptosis in MDA-MB-231 cells while it was unresponsive in the MCF-7 cells. Further, our in silico results revealed significant interaction and better inhibition of erysolin to CDK2, CDK6, Bcl2 and ER-α compared to the standard reference compounds. Moreover, erysolin also showed higher affinity against interface of p53-MDM2 which might stimulate p53 in tumor cells or induces restoration of mutant p53 thereby making these cancer cells to undergo apoptosis. Feeble interactions with pro-apoptotic protein BAX, caspases 3 and 8 also indicate possible involvement of extrinsic pathway in the erysolin-mediated apoptosis. The results showed that erysolin is a promising natural anticancer and anti-estrogenic agent. Further, the in silico protocol for elucidating and validating apoptosis mechanism(s) by unknown compounds could also be used.
Keywords: Erysolin, Apoptosis, Molecular Docking, MCF-7, MDA-MBA-231
Cite this paper: Fahad M. Al-Khodairy, M. Kalim A. Khan, M. Kunhi, Manogaran S. Pulicat, Salman Akhtar, Jamal M. Arif, In Silico Prediction of Mechanism of Erysolin-induced Apoptosis in Human Breast Cancer Cell Lines, American Journal of Bioinformatics Research, Vol. 3 No. 3, 2013, pp. 62-71. doi: 10.5923/j.bioinformatics.20130303.03.
Article Outline
1. Introduction
- Novel chemopreventive and anticancer therapeutic approaches are continuously in the limelight in a quest for safer and effective drug candidates. Numerous molecular targets of various cellular processes during tumor development have been already tested with different classes of natural and synthetic compounds. However, the immortality of cancer cells has lured numerous scientists to study possible ways to induce apoptosis in cancer cells for developing novel anticancer drugs. Numerous novel compounds and anticancer drugs are able to significantly and sometimes specifically induce apoptosis in variety of cancer cells[1, 2].In the past several decades, p53 has gained special status in the prevention of cancer due to its ability to act in many cellular processes including cell-cycle checkpoints, DNA repair, senescence, angiogenesis, and apoptosis[3]. It is this ability of p53 which makes it as a soft target in many cancers for frequent mutations subsequently making the tumorigenesis to progress[4, 5]. Although p53 stimulates a wide network of signals that acts through two major apoptotic pathways (extrinsic and intrinsic) but several studies suggested that these pathways may be converging rather than distinct pathways[6].Since one of the most important functions of p53 is its ability to activate apoptosis, therefore, manipulation of the apoptotic functions of p53 constitutes an attractive target for cancer therapy. The antitumor function of p53 is often attenuated or even omitted mainly due to two alternative mechanisms, direct gene alterations in p53 or negative control by MDM2 protein. The MDM2 genes are found to be abnormally up-regulated in several human cancers[7]. Over-expression of MDM2 promotes cell growth and accelerates tumorigenesis[8, 9]. Hence a number of strategies have been used to define small molecules binding at the interface between the two proteins interfering with the p53-MDM2 interaction by possible blocking of MDM2 expression, inhibiting MDM2 ubiquitin ligase activity and/or blocking p53-MDM2 interaction. This restricts the activity of MDM2 protein and allows p53 to function as a normal tumor suppressor[10]. Several peptidic, non-peptidic and glycoalkaloid molecules able to bind at the p53-MDM2 transactivation domain-binding cleft may consequently block the p53-MDM2 interaction[11, 12, Akhtar S. PhD Thesis June 2012]. In contrast, Issaeva et al., 2004[13] reported the small molecule RITA, which binds to p53 and targets it for proteasomal degradation. A few of the potent and selective agents (JNJ-26854165, RG7112) have also been advanced to clinical trials for the treatment of cancer[10, 14]. In recent years, rocket plant (Eruca sativa) has gained greater importance as a vegetable and spice, especially among Europeans. Erysolin, a naturally occurring sulfonyl analogue of sulforaphane containing -N=C=S moiety (Fig.1) was originally extracted from rocket plant. Erysolin is a scarcely studied natural compound though it has been a potent inducer of apoptosis in human colon cancer cells[15], inducer of caspase 8[15], anti-genotoxic and chemopreventive agent in human hepatoma HepG2 cells towards benzo[a]pyrene[16], and induced reactive oxygen species-mediated arsenic trioxide cytotoxicity in human leukemic cells[17]. However, the mechanism(s) of erysolin-mediated apoptosis in human cancers including breast cancer have not been elucidated.
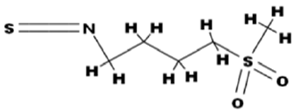 | Figure 1. Chemical structure of erysolin |
2. Materials and Methods
2.1. Cell Lines
- Human breast carcinoma cell lines (MCF-7, MDA-MB-231) were purchased from American Type Culture Collection (Rockville, MD, USA). All the cell culture reagents and media were obtained commercially from Sigma-Aldrich (St. Louis, MO, USA). A Vybrant apoptosis assay kit #2 was purchased from Molecular Probes, Inc. (Eugene, OR, USA).
2.2. Treatment of Cells and Apoptosis
- MCF-7 and MDA-MB-231 cells were cultured in RPMI1640 medium using 10% fetal calf serum to 80-90% confluence, then subcultured using 60 mm dishes and treated with 10 and 50 μM of erysolin (Sigma-Aldrich, St Louis, MO, USA), dissolved in DMSO (final conc. 0.1%), for 72 h at 37°C in the CO2 incubator under 5% carbon dioxide. The experimental conditions were pre-determined in preliminary dose- and time-dependent experiments.The cells were harvested by trypsinization and cells were washed in cold phosphate-buffered saline for analysis of apoptosis as per the Manufacturer’s protocol. The cells were centrifuged followed by staining with annexin V and propidium iodide in annexin-binding buffer. After 15 min incubation at room temperature, the fluorescence was measured using a flow cytometer (FACScan, Becton Dickenson, USA). The results were analyzed using CellQuest Pro software and represented as percentage of normal (viable) and apoptotic cells.
2.3. Docking Simulations
- Docking simulation was performed using the AutoDock Tools 4.0 in order to find the preferred binding conformations of ligands in the receptor[22]. Docking to macromolecule was performed using an empirical free energy function and Lamarckian Genetic Algorithm, with an initial population of 250 randomly placed individuals, a maximum number of 106 energy evaluations, a mutation rate of 0.02, and a crossover rate of 0.80. One hundred independent docking runs were performed for each ligand. Results differing by <2.0 Å in positional root mean-square deviation (RMSD) were clustered together and represented by the result with the most favorable free energy of binding[23].
2.4. Ligand Preparation
- Erysolin (C6H11NO2S2) (CID: 3080557) was searched on PubChem database. The PubChem database contains structural and functional information about different organic compounds. The structure of a compound is summarized as its SMILES string. This string was taken for the ligand of interest and submitted to another online software Babel Molecular Structure Format convertor(http://www.vcclab.org/lab/babel/). It takes SMILES string as input and generates 3D structure of the molecule whose structural coordinates file can be downloaded in PDB format suitable for AutoDock Tools 4.0. Energy minimization was carried out by applying cff force field[24].
2.5. Protein Preparation
- The crystal structure of proteins (Table 1) taken in this study were extracted from Brookhaven Protein Data Bank (http://www.rcsb.org/pdb). Proteins were prepared for docking in such a way that all the nonreceptor atoms such as water, ions, etc. were removed. Kollman charges were assigned[25-27]. Solvation parameters were added to the final macromolecule structure using the Addsol utility of AutoDock. By applying CharMM force field[28] energy minimization was carried out to remove the bad steric clashes using steepest descent algorithm for 1000 steps at RMS gradient of 0.01.
2.6. Validation of Docking Method
- To ensure that the ligand orientations and positions obtained from the docking studies were likely to represent valid and reasonable potential binding modes of the inhibitors, the docking methods and parameters used were validated by redocking experiments. Each ligand was docked into the native protein to determine the ability of AutoDock program to reproduce the orientation and position of the ligand observed in the crystal structure. The top ranking conformational clusters from this dock were evaluated in terms of RMSD between docked position and experimentally determined position for the ligand. The low RMS (1.35 Å) between the experimental and docked co-ordinates of ligand indicated same binding orientation that favored the validation of docking method (Fig. 2).
|
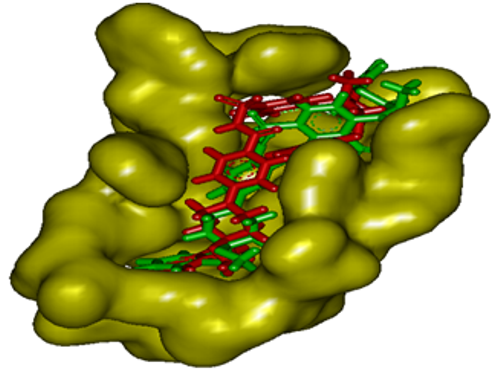 | Figure 2. Validation of docking method by superimposing the inhibitor present in the crystal structure of Bcl2 (red) and that after redocking the same (green) with AutoDock Tool 4.0 |
2.7. Drug-likeness, ADME & Toxicity Prediction Studies
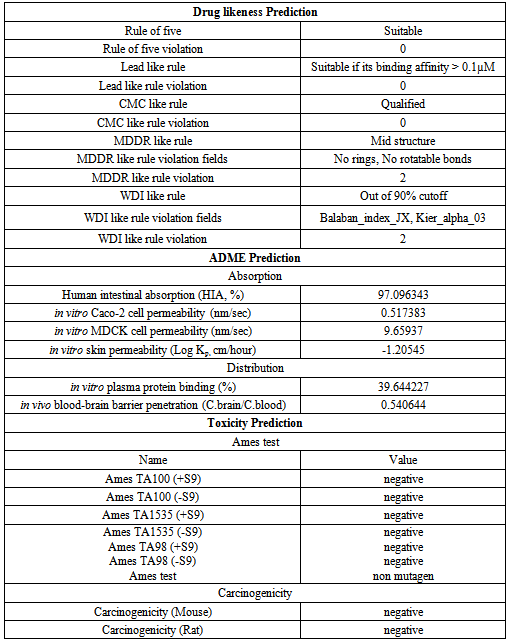
- The ligands with good pharmacological and drug-likeness properties are very crucial for structure based drug discovery. The absorption, distribution, metabolism, excretion and toxicity (ADMET) are the most important part of pharmacological studies of lead molecules and these can be predicted by computational biology tools. Hence, erysolin was tested for its drug-likeliness, ADME profile and toxicity analysis through open source tool PreADMET (http://preadmet.bmdrc.org). The ADME includes the extent and rate of absorption, distribution, metabolism and excretion. Pre-ADMET uses Caco2-cell (heterogeneous human epithelial colorectal adenocarcinoma cell lines) and MDCK (Madin-Darby Canine Kidney) cell models for oral drug absorption prediction and skin permeability, and human intestinal absorption model for oral and transdermal drug absorption prediction. Similarly, the program uses BBB (blood brain barrier) penetration and plasma protein binding models to predict the distribution. Pre-ADMET also predicts toxicity based on the mutagenicity of Ames parameters and rodent carcinogenicity assays of rat and mouse[29-43].
2.8. Statistical Analysis
- All the in vitro experiments with the cell lines were performed in triplicate and the values expressed as mean ± SD. The statistical significance was calculated by student’s t-test and p<0.05 was considered to be significant.
3. Results and Discussion
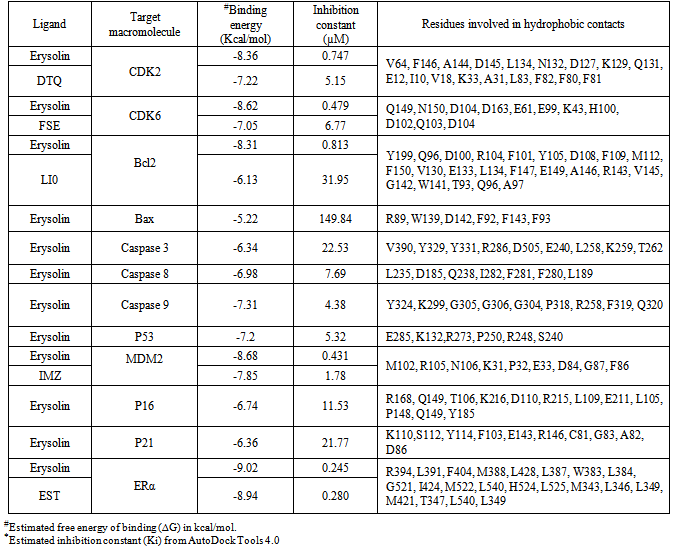
- Resistance to apoptosis is undoubtedly one of the major causes behind the immortality of cancer cells. There have been intense search of natural or synthetic compounds capable of inducing substantial apoptosis in the cancer cells for development of anticancer drugs. Our in vitro study showed that erysolin (10 µM) neither induced apoptosis nor cytotoxicity in the MCF-7 cells (p53 wild type and ER-α positive) in 72 h incubation while the p53 mutant and ER-α negative MDA-MB-231 cells showed induction of moderate apoptosis of 20% (Fig. 3). Increasing the dose of erysolin in the incubation medium to 50 µM resulted in significant increase in apoptosis in MDA-MB-231 cells (70%) compared to the MCF-7 cells (50%) (p<0.05). Further, erysolin (50 µM) also resulted in some necrosis in the MCF-7 cells. Based on our previous studies[18-21], we tried to elucidate the mechanism of erysolin-induced apoptosis in the cell lines by an in silico approach using various molecular targets of apoptosis pathway.The most perceptive relationship between p53-mediated transactivation and apoptosis is its ability to regulate transcription of pro-apoptotic members of the Bcl-2 family; however, the increased ratio of pro- to anti-apoptotic Bcl-2 proteins favors the release of apoptogenic proteins from the mitochondria, caspase activation, and apoptosis. MDM2 negatively regulates p53 in three different ways. First, by binding to p53 transactivation domain and inhibits its transcriptional activity[44]; second, MDM2 act as ubiquitin ligase promotes p53 degradation[45, 46]; and third, by binding to p53 and its expulsion from nucleus[47]. Therefore, in the presence of MDM2, p53 protein is inactivated and does not stimulate the expression of genes involved in apoptosis, cell cycle arrest or DNA repair. Imidazoline, a well known inhibitor binds to MDM2 in the p53-binding pocket and activates the p53 pathway in cancer cells thereby causes cell cycle arrest and apoptosis[48]. Our docking simulations revealed that erysolin showed relatively stronger binding affinity to MDM2 in terms of binding energy -8.68 kcal/mol and inhibition constant (Ki) 0.431 µM as compared to its known inhibitor imidazoline having binding energy -7.85 and Ki 1.78 µM (Fig. 4A and Table 2). Furthermore, M102, R105, N106, K31, P32, E33, D84, G87 and F86 are the common residues involved in interaction of both erysolin and imidazoline with MDM2. The inhibition of MDM2 by erysolin may activate the p53 pathway and as a possible consequence shall activate apoptosis. Molecular interaction of erysolin with p53 alone showed relatively less significant and weak interaction when compared to MDM2 as it showed less binding energy (-7.2 kcal/mol) with higher Ki of 5.32 µM (Fig. 4B and Table 2). This decrease in the binding energy and increase in Ki value can be directly correlated with the fact that erysolin preferred to bind MDM2 with greater affinity in comparison to p53. The docked complex of erysolin was surrounded by the E285, K132, R273, P250, R248, S240 key amino acid residues. The results obtained with p53 certainly propose erysolin as better antagonist of MDM2 as compared to p53 in a process to block the degradation of normal p53 protein and thus releasing it free to induce apoptosis. This result further strengthens our in vitro data with MCF-7 cells indicating that erysolin possibly induces p53-mediated apoptosis by releasing and stabilizing the p53 bound to MDM2. However, apoptosis seen in the MDA-MB-231 cells at lower concentration (10 µM) of erysolin might be due to the fact that erysolin possibly initiate the expression of p53 or restore the active p53 from mutant form of p53. The substantial increase in apoptosis at 50 µM erysolin could be attributed to its higher binding with MDM2 thereby making newly expressed p53 or restoring mutant p53 to active form to participate in the execution of apoptosis. However, further evidences from the wet lab experiments are needed to strengthen this hypothesis.In continuation, we also studied the interaction of erysolin with the p21WAF1/CIP1, a downstream target of p53 and a CDK inhibitor which is activated following DNA damage to induce cell-cycle arrest, allowing the cell time to repair its damaged genome and to prevent apoptosis[49, 50]. Though commonly associated with a growth inhibitory role, p21WAF1/CIP1 can also function in cell proliferation. It has also been shown that p21WAF1/CIP1 deficient thymocytes undergo rapid apoptosis following damage. Inhibiting the levels of p21WAF1/CIP1 diminishes its anti-apoptotic effect on cells and makes cells more amenable to cell death[51]. Molecular docking of erysolin with p21WAF1/CIP1 showed lesser binding affinity with binding energy of -6.36 kcal/mol and Ki value of 21.77 µM. K110, S112, Y114, F103, E143, R146, C81, G83, A82, D86 are the key residues involved in hydrophobic interaction (Fig. 4C and Table 2). Thus, inhibiting the activity of p21WAF1/CIP1 by erysolin may induce apoptosis in cancer cells.P16 acts as a negative regulator of cell cycle by binding to and inhibiting cyclin-dependent kinases rendering the retinoblastoma protein inactive. This effect blocks the transcription of important cell cycle regulatory proteins and results in cell cycle arrest by subsequently inducing apoptosis. Docking simulations with p16 revealed feeble interactions of erysolin with binding energy of -6.74 kcal/mol and Ki value of 11.53 µM (Fig. 4D and Table 2). R168, Q149, T106, K216, D110, R215, L109, E211, L105, P148, Q149, Y185 residues are involved in molecular interaction. Feeble interactions of erysolin with p21WAF1/CIP1 and p16 further promote that erysolin is a potent inducer of apoptosis by inhibiting the p53-MDM2 complex formation.
|
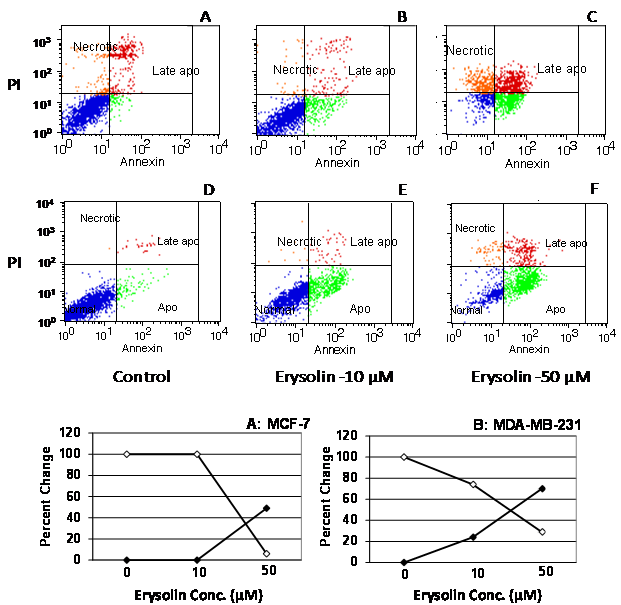 | Figure 3. Apoptosis and cell viability in MCF-7 and MDA-MB-231 incubated with erysolin (10 and 50 µM) for 72 h. The cells were harvested and analyzed by FACS and Cell Quest Pro software. The data was normalized against the vehicle control (100% for viability or 0% for apoptosis). The values are mean of 3 replicates. Panels A-C represents the flow cytometric maps from MCF-7 while panels D-F are from MDA-MB-231. The solid circles line in the panels A: MCF-7 and B: MDA-MB-231 showed % apoptosis while hollow circles line represented % viability |
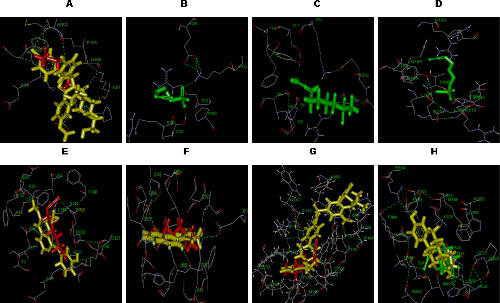 | Figure 4. Visualization of docked structure in PyMol. Panel A: Docked complex of MDM2 with erysolin (red) and ligand IMZ (yellow). Panel B: Docked complex of p53 with erysolin (green). Panel C: Docked complex of p21 with erysolin (green). Panel D: Docked complex of p16 with erysolin (green). Panel E: Docked complex of CDK2 with erysolin (red) and ligand DTQ (yellow). Panel F: Docked complex of CDK6 with erysolin (red) and ligand FSE (yellow). Panel G: Docked complex of Bcl-2 with erysolin (red) and ligand LI0 (yellow). Panel H: Docked complex of ER-α with erysolin (green) and ligand EST (yellow). Hydrogen bonds are shown by green dotted lines providing stability to the docked complexes |
|
4. Conclusions
- It can be extracted from the results that erysolin-induced apoptosis in both MCF-7 and MDA-MB-231 cells by inhibiting the p53-MDM2 interaction and Bcl2 with subsequent activation of BAX, caspases 8 and 3 thereby indicating involvement of p53-mediated extrinsic pathway of apoptosis. Furthermore, strong affinity binding of erysolin with the active sites of CDK2 and CDK6 subsequently leads to the induction of apoptosis. In addition, erysolin has also shown strong anti-estrogenic activity in the in silico docking simulations indicating that it might be developed as an effective anti-estrogenic agent in the treatment of ER-positive breast cancers. Our in silico data has again complemented the in vitro data with possible underlying mechanism(s) of action[18, 20, 21, 65] hence in silico protocol could be used in the mechanism-based screening of new compounds against cancers and other diseases by using disease-specific molecular and cellular targets.
ACKNOWLEDGEMENTS
- The cell culture studies were performed at the KFSH&RC, Riyadh, KSA while the bioinformatics experiments were done at the Integral University, Lucknow, India. Thanks to Dr. M. Kuddus for formatting the manuscript. The authors are also thankful to the Dr. Sultan Al-Sedairy, Executive Director (KFSH&RC) and Prof. S.W. Akhtar for their infrastructure support to carry out this study.