-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2013; 3(3): 42-61
doi:10.5923/j.bioinformatics.20130303.02
Sequence, Structural and Functional Characterization of Homogentisate-1,2-dioxygenase of homo sapiens: An in silico Analysis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSen Gupta Parth Sarthi, Banerjee Shyamashree, Bandyopadhyay Amal Kumar
Department of Biotechnology, The University of Burdwan, Burdwan, West Bengal, 713104, India
Correspondence to: Bandyopadhyay Amal Kumar, Department of Biotechnology, The University of Burdwan, Burdwan, West Bengal, 713104, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In silico characterization and molecular modeling of a protein opens wide scope for the prediction of structural and functional information. It is most significant and helpful when very little information about three dimensional (3D) structure of protein(uncharacterized) available in the Protein Data Bank (PDB). So, the present study has been undertaken to carried out in silico prediction of structure and function of Homogentisate-1,2-dioxygenase of homo sapiens. Primary structure analysis reveals that all the six sequences of Homogentisate-1,2-dioxygenase arehydrophobic in nature (due to the high content of non- polar residues) without transmembrane region. The aliphatic index computation infers that Homogentisate-1,2-dioxygenase of homo sapiens can’t tolerate wide range of temperature. Secondary structure analysis shows that all of the sequences of Homogentisate-1,2-dioxygenase have predominant sheet and random coil structure. Sub cellular localization prediction suggested that all of the sequences are secretory without transit peptide. 3D structure of the protein is predicted and characterized. Energy minimization, Refinement and validation of the structure is done, which suggest the structure to be a very good quality. The model generated for Homogentisate-1,2-dioxygenase is successfully submitted to the Protein Model Database (PMDB) having PMID PM0079008. Active site of the structure, Protein disorder, disorder propensity and Average Area Buried Upon Folding is also predicted and analyzed. Fold of the protein and motif of the protein is also predicted. The function of the protein is predicted and analyzed. This study highlights the sequence, structural and functional information of the protein.
Keywords: Homo Sapiens, Molecular Modeling, Homogentisate-1,2-dioxygenase, Disorder, Aliphatic Index
Cite this paper: Sen Gupta Parth Sarthi, Banerjee Shyamashree, Bandyopadhyay Amal Kumar, Sequence, Structural and Functional Characterization of Homogentisate-1,2-dioxygenase of homo sapiens: An in silico Analysis, American Journal of Bioinformatics Research, Vol. 3 No. 3, 2013, pp. 42-61. doi: 10.5923/j.bioinformatics.20130303.02.
Article Outline
1. Introduction
- Homogentisate-1,2-dioxygenase (HGD) is an iron containing enzyme which catalyzes the conversion of homogentisate to 4-maleylacetoacetate. An absence or deficiency of homogentisate-1,2-dioxygenase will result in alkaptonuria (AKU). Homogentisate 1,2-dioxygenase is involved in the catabolism of aromatic rings, more specifically in the breakdown of the amino acids tyrosine and phenylalanine[1].HGD appears in the metabolic pathway of tyrosine and phenylalanine degradation when homogentisate is produced. Homogentisate reacts with HGD to produce maleylacetoacetate, which then is further used in the metabolic pathway. HGD requires the use of Fe2+ and O2 in order to cleave the aromatic ring of homogentisate[2]. AKU is due to the inability of the body to deal with homogentisate, which when oxidized by the body will produce the compound known as the ochronotic pigment. This first of these effects is that the patient’s earwax will begin to turn black or red, depends on the patient’s diet, since the blood becomes oxidized and thus turns black due to excess of the ochronotic pigment. The other effect of the ochronotic pigment is that it can accumulate in the body’s connective tissue leading to degenerative arthritis, as the person grows older[2]. The active site of Homogentisate 1,2-dioxygenase was determined through the crystal structure, which was captured through the work of Titus et al[1]. Borowski et al. propose a mechanism for HGD which is The opening of the aromatic ring in homogentisate is a multi-step process. In the first two steps Fe2+ coordinates to the carbonyl and ortho phenol oxygens. The iron atom is also coordinated to His335, His371, and Glu341. O2 then binds to the iron atom[2], subsequently reacting with the aromatic ring to form a peroxo-bridged intermediate. In the next step, O2 is cleaved with the formation of an epoxide. This epoxide intermediate allowing radical reactions to eventually open and oxidize the six-membered ring.Plenty of work has been done on Homogentisate - 1, 2 -dioxygenase of various organisms[3 to 13].The Protein Data Bank (PDB) (www.rscb.org) contain very little information about three dimensional structure of Homogentisate - 1, 2 - dioxygenase of Homo sapiens. Therefore, it is very interesting as well as important to carry out the study of Homogentisate-1,2-dioxygenase of Homo sapiens. In the present study Sequence, Structural and Functional Characterization of Homogentisate-1,2-dioxygenase of Homo sapiens: An in silico analysis. We used various servers and softwares to analyze the sequences and to predict the three dimensional structure and function of the Homogentisate-1,2-dioxygenase. The predicted 3D structure is also successfully submitted in the Protein Model Data Base (PMDB)[14] having PMID PM0079008. The results of this work can further help researcher by providing theoretical basis on enzymological properties, structure and function of Homogentisate-1,2-dioxygenase in future.
2. Materials and Methods
2.1. Sequence Retrieval
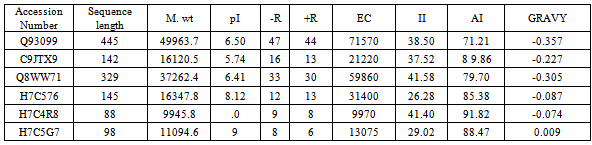
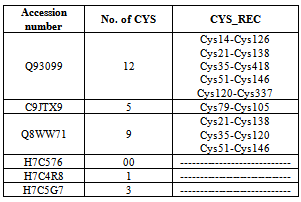
- A total of six sequences of Homogentisate - 1, 2 - dioxygenase protein were retrieved from the manually curate public protein database Swiss-Prot[15]. Swiss - Prot is scanned for the keyword Homogentisate-1,2-dioxygenase and Homo sapiens. The search result yielded 6 sequences of Homogentisate-1,2-dioxygenase (HGD) gene family of Homo sapiens (Table 1). All of the six sequences were retrieved in FASTA format and used for further analysis.
|
2.2. Primary Structure Analysis Using Computational Tools and Servers
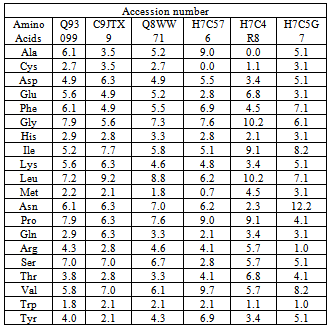
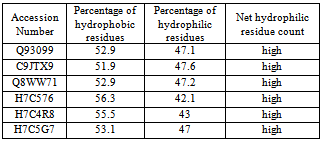
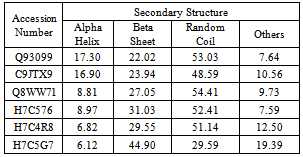
- The amino acid composition (table 2) of Homogentisate - 1, 2-dioxygenase sequences were computed using the tool CLC free Workbench[http://www.clcbio.com/products/clc-sequence-viewer/]. Percentages of hydrophobic and hydrophilic residues were calculated from the primary structure analysis and tabulated in table 3.
|
|
|
|
|
2.3. Template Selection and Molecular Homology Modeling
- The protein sequence of Homogentisate-1,2-dioxygenase having accession ID Q8WW71 has no three dimensional structure present in Protein Data Bank (PDB).Therefore, the template (PDB ID 1F2V_A ) were identified by the BLASTP (http://www.ncbi.nlm.nih.gov:80 /BLAST/) analysis in the Protein Data Bank (PDB). Three dimensional structure of Homogentisate - 1, 2 - dioxygenase having accession ID Q8WW71 was generated using ESyPred3D (http:// fundp. ac. be / urbm/bioinfo/esypred/) automated homology modeling server predicted the homology model based on a package MODELLER and by using Swiss Model[19].
2.4. Energy Minimization, Model Evaluation and Submission
- The energy minimization for the 3D structure was carried out using NAMD[20] utilizing CHARMM force field and NOMAD-Ref server[21] which utilizes Gromacs forcefield according to steepest descent, conjugate gradient and L - BFGS methods. Further, refinement of the modeled structure was performed using 3 Drefine server[22]. Structural evaluation, validation and stereochemical analyses were performed using various evaluation tools such as Rampage , Procheck[23], Errat[24], Ramachandran plot 2[25], and Vadar[26]. Protein Quality was checked by Resprox[37]. Furthermore, visualization and analyses of the generated model was performed using UCSF Chimera 1.5.3. The generated 3D model was successfully submitted in the Protein Model Data Base (PMDB) having PMID PM0079008. PMDB Protein Model Database, which collects three dimensional protein models obtained by structure prediction methods. Users can both contribute new models and search for existing ones. The database currently stores all models submitted to the last four editions of the CASP experiment.
2.5. Active Site Prediction
- After the final model was built, the active site of the structure was predicted using the tool Meta pocket 2(38).The visualization and analysis of the active site was performed by using UCSF Chimera 1.5.3.
2.6. Identification of Motif, Fold and Functional Domain
- Motif of the protein were identified using the tools Prosite [29] and MotifScan[30].The protein fold was predicted using the software PFP-pred[31].
2.7. Function Prediction
- The function of the protein Homogentisate - 1, 2 - dioxygenase of Homo sapiens was predicted by using ProFunc 2[32]. The aim of the ProFunc server is to help identify the likely biochemical function of a protein from its three-dimensional structure. It uses a series of methods, including fold matching, residue conservation, surface cleft analysis, and functional 3D templates, to identify both the protein's likely active site and possible homologues in the PDB.
3. Result and Discussion
3.1. Primary Structure Analysis
- The results of primary structure analysis suggest that all of the sequences of Homogentisate-1,2-dioxygenase are hydrophobic in nature due to the presence of high content of non- polar residues (tables 2 and 3). The kye-dollitle hydrophobicity is analyzed using ProtScale and shown in figure 1.
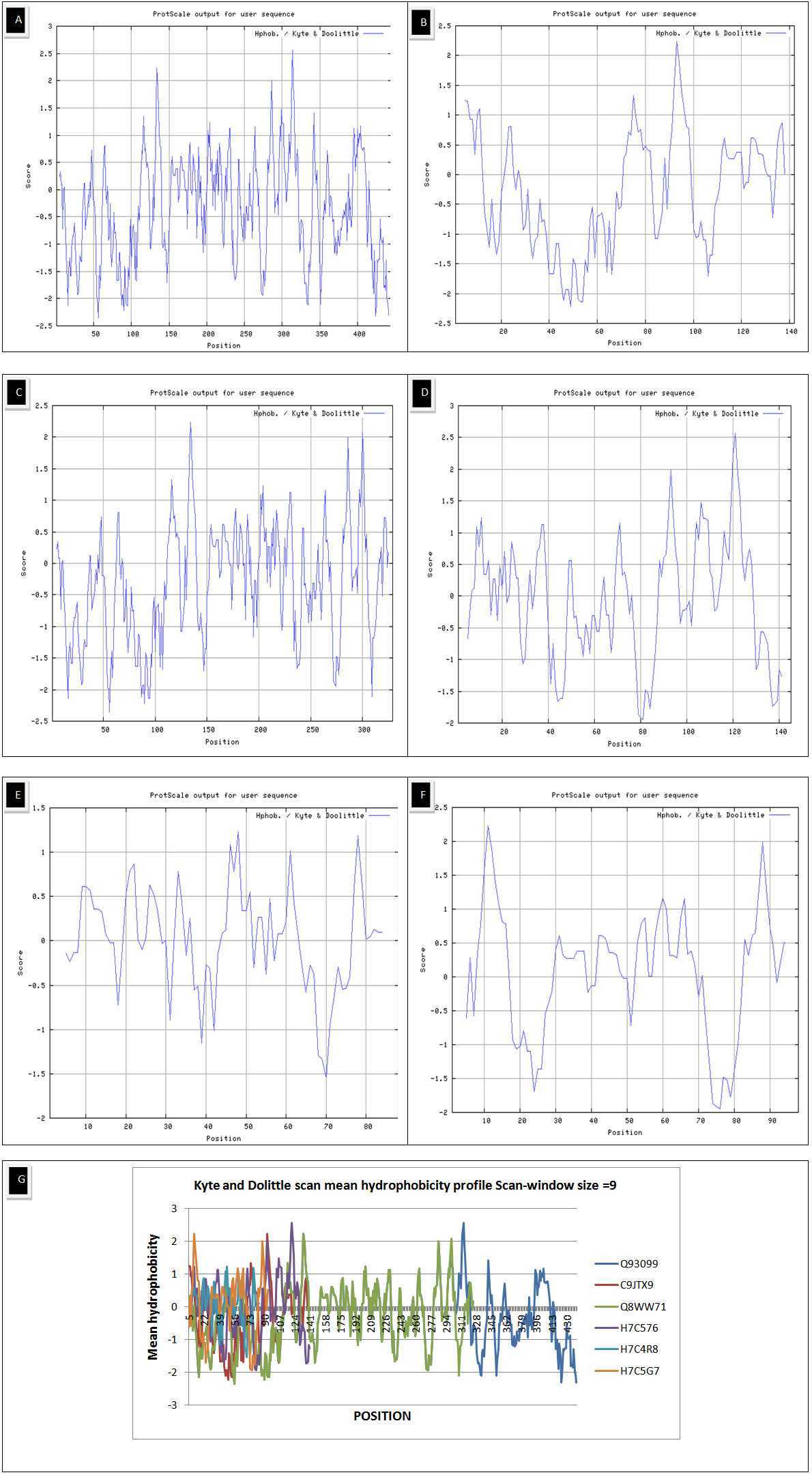 | Figure 1. Kyte-doolittle hydrophobicity by ProtScale. (A) For the sequence with accession id Q93099 (B) For the sequence with accession id C9JTX9 (C) For the sequence with accession id Q8WW71 (D) For the sequence with accession id H7C576 (E) For the sequence with accession id H7C4R8 (F) For the sequence with accession id H7C5G7 (G) Kyte-Doolittle scale mean hydrophobicity profile for all the four sequences of Homogentisate-1,2-dioxygenase |
|
3.2. Molecular Homology Modeling, Energy Minimization and Model Evaluation
- The protein sequence of Homogentisate-1,2-dioxygenase having accession ID Q8WW71 has no three dimensional structure present in Protein Data Bank (PDB).Therefore, the template (PDB ID 1EYB_A ) are identified by the BLASTP analysis in the Protein Data Bank (PDB)(Table 8).
|
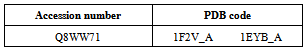
 | Figure 2. Disordered Regions of the Chalcone Synthase protein (A) Disorder Propensity Sum (B) schematic diagram of disorder Probability over the entire sequence |
|

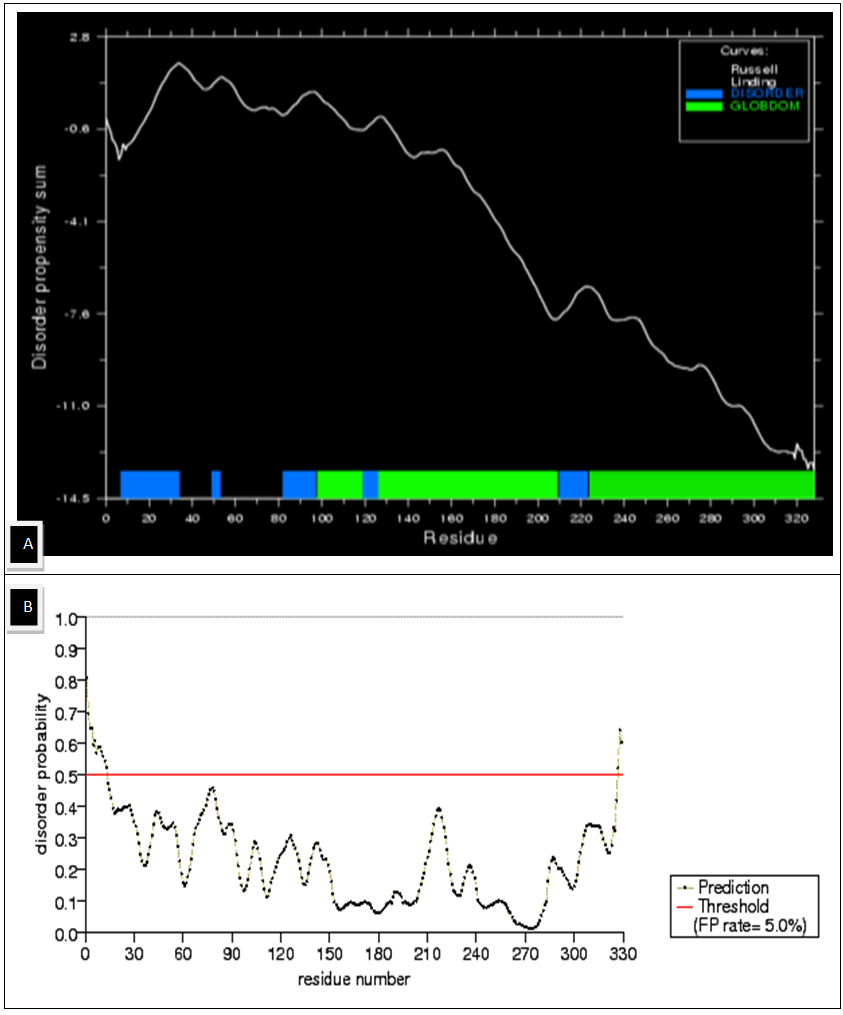
 | Figure 3. Average Area Buried upon Folding For all the Amino acids residue of the Protein predicted by Protscale from ExPASy |
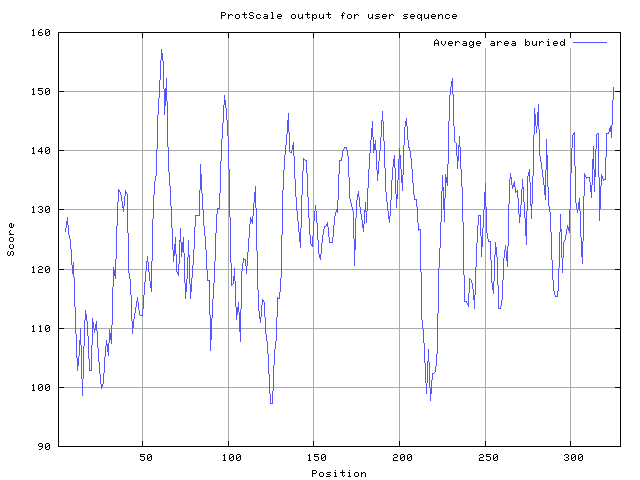
 | Figure 4. Ramachandran plot using (A)Rampage (B)Vadar (C)Ramachandran Plot 2 (D, E) Procheck (F) Z score using Errat |
|

|
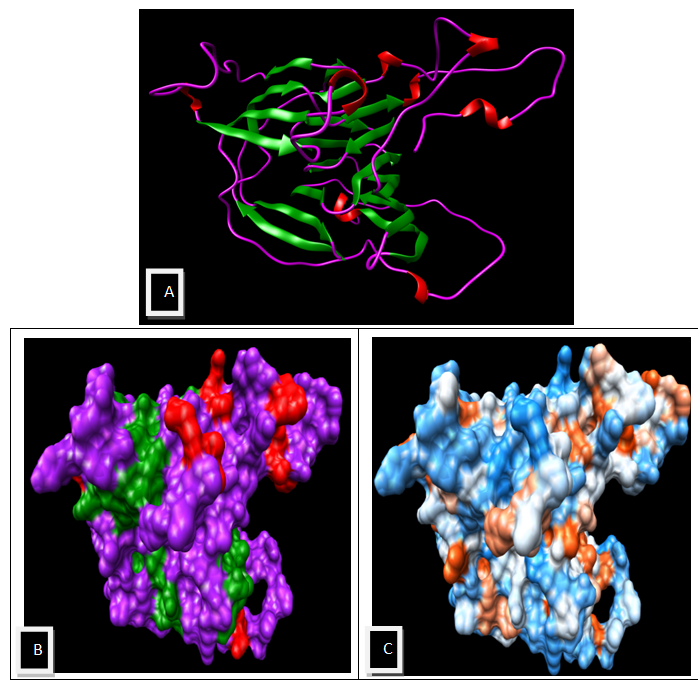 | Figure 5. Visualization by using UCSF Chimera 1.5.3 (A) Secondary Structure of the Model (B) Surface View of the model Structure (C) Molecular Surface Colored by amino acid Hydrophobicity (Dodger Blue-most hydrophilic, White- Moderate, Orange Red- most hydrophobic) |
3.3. Characterization and Active Site Prediction of the Structure
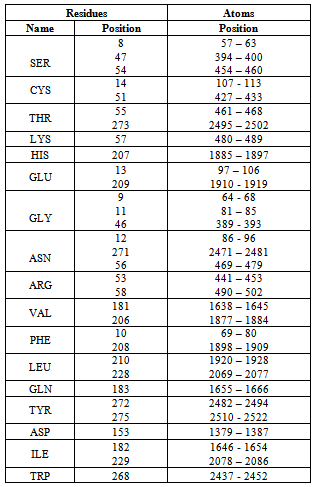
- The modeled structure is characterized by their secondary structure contents and it is found that Homogentisate - 1, 2 - dioxygenase contains high amount of alpha helix and coils (table 13) in respect to beta sheet just like the primary structure analysis (table 6).The secondary and topological three dimensional structure is calculated using PDBsum and shown in figure 6.The total protein intrinsic disorder and the protein disorder propensity was detected and calculation shows that protein is mostly maintaining the ordered structure except one place at the middle and at the N terminal and C terminal region also shown in the modeled structure ( Figure 7).For more information just follow this link http:// www. ebi.ac.uk/pdbsum/ by using PDB code: gl39 and Password: 203313 or directly by bookmarking this link: http:/ /www. ebi.ac.uk/thornton-srv/databases/cgi- bin/ pdbsum/ GetPage. pl?pdbcode =gl39 & code =203313 .The visualization and analysis of the active site was performed by using UCSF Chimera 1.5.3 (Figure 8).The active site of the structure is analyzed using the tool Meta pocket 2 (Table 14).
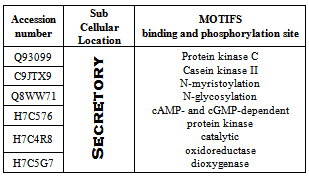
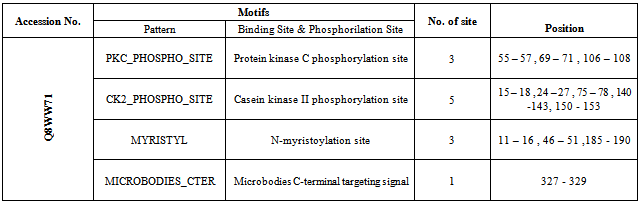
3.4. Identification of Motif, Fold, Functional Domain and Function Prediction
- Motif of the protein is identified using the tools Prosite and MotifScan (Table 16). PFP-pred predicted the protein fold type to be Small inhibitors, toxins, lectins (Table 15).The function of the protein is identified using ProFunc 2 and it predicted that the Homogentisate-1,2-dioxygenase is involved in many biological and catalytical functions (Table 17).For more information related to function follow the link to ProFunc home pagehttp://www.ebi.ac.uk/thornton-srv/databases/profunc using PDB code: gl39 and Password: 203313 or directly by bookmarking this link: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/profunc/GetResults.pl?source=profunc&user_id=gl39&code=203313.
|

 | Figure 6. (A) Secondary structure of the Modeled Structure, Helices are labeled as H; Beta turn as β; Gamma turn as χ and Beta hairpin as ⥰ (B) Topology of Secondary Structure of Model, Helices and Sheets are shown in Pink and Red |
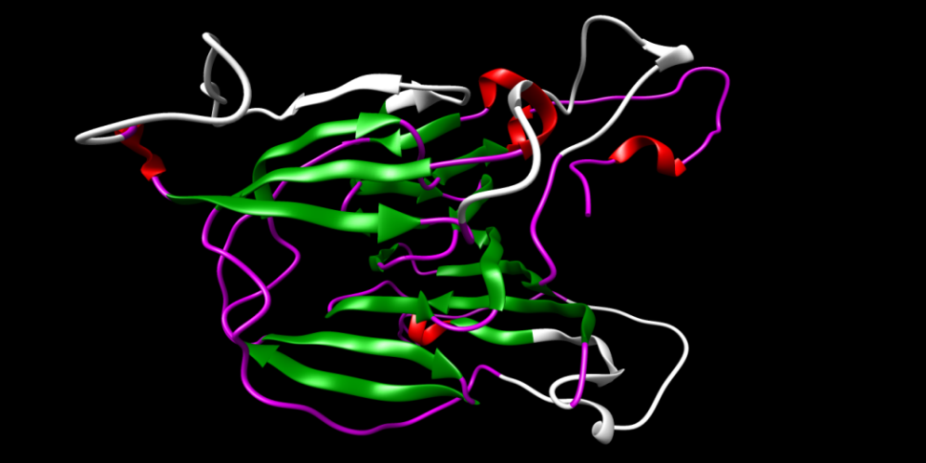 | Figure 7. The total protein intrinsic disorder is shown in the structure (white color) |
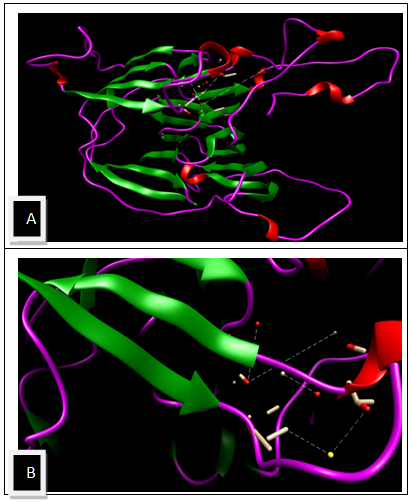 | Figure 8. The active site predicted using metapocket 2 A) active site in the modeled structure B) Enlarge view of the active site |
|
|

|
|

4. Conclusions
- The sequences of homogentisate-1,2-dioxygenase from Homo sapiens are chosen for the present study as because very little or no information of the protein is present currently. Primary analysis suggests that it is hydrophobic in nature and localized as secretory without transit peptide. Physico-chemical characterization provides essential information or data about the protein and its properties. Secondary structure analysis suggests that the protein is dominantly of sheet and random coil. It has no transmembrane region and highly structured as shown by flexibility and disordered studies. Active site analysis suggests that the structure has some highly conserved residues. Identification of motifs showed that it has many binding and phosphorylation site indicating the engagement of the protein in many catalytical processes. Functional analysis suggests that it is involved in protein binding and dimerization activity as well as many other biological functions. The present study provides all the necessary information about sequence, structure and function of the protein to the scientist for the further research in future.
ACKNOWLEDGEMENTS
- Authors are grateful to the Department of Biotechnology (DBT), Govt. of India for the computational facility laboratory.
References
| [1] | Titus, Greg P; Mueller, HA; Burgner, J; Rodríguez De Córdoba, S; Peñalva, MA; Timm, DE (2000). "Crystal structure of human homogentisate dioxygenase". Nature Structural Biology 7 (7): 542–546. doi:10.1038/76756. PMID 10876237. |
| [2] | Borowski, Tomasz; Georgiev, V; Siegbahn, PE (2005). "Catalytic Reaction Mechanism of Homogentisate Dioxygenase: A Hybrid DFT Study". Journal of American Chemical Society 127 (49): 17303–17314. doi:10. 1021/ ja054433j. PMID 16332080 |
| [3] | Preston AJ, Keenan CM, Sutherland H, Wilson PJ, Wlodarski B, Taylor AM, Williams DP, Ranganath LR, Gallagher JA, Jarvis JC. Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone. Ann Rheum Dis. 2013 Mar 19. |
| [4] | Yang YJ, Guo JH, Chen WJ, Zhao R, Tang JS, Meng XH, Zhao L, Tu M, He XY, Wu LQ, Zhu YM. First report of HGD mutations in a Chinese with alkaptonuria. Gene. 2013 Apr 15;518(2):467-9. doi: 10.1016/j.gene.2013.01.020. Epub 2013 Jan 24. |
| [5] | Hu M, Ma HW, Luo Y, Wang L, Song Y, Li F.[Gene diagnosis of alkaptonuria in an infant]. Zhongguo Dang Dai Er Ke Za Zhi. 2012 Oct;14(10):796-7. |
| [6] | Millucci L, Spreafico A, Tinti L, Braconi D, Ghezzi L, Paccagnini E, Bernardini G, Amato L, Laschi M, Selvi E, Galeazzi M, Mannoni A, Benucci M, Lupetti P, Chellini F, Orlandini M, Santucci A. Alkaptonuria is a novel human secondary amyloidogenic disease. Biochim Biophys Acta. 2012 Nov;1822(11):1682-91. doi: 10. 1016 /j. bbadis. 2012.07.011. Epub 2012 Jul 28. |
| [7] | Magesh R, George Priya Doss C. Computational methods to work as first-pass filter in deleterious SNP analysis of alkaptonuria. ScientificWorldJournal. 2012;2012:738423. doi: 10.1100/2012/738423. Epub 2012 Apr 19 |
| [8] | Taylor AM, Preston AJ, Paulk NK, Sutherland H, Keenan CM, Wilson PJ, Wlodarski B, Grompe M, Ranganath LR, Gallagher JA, Jarvis JC. Ochronosis in a murine model of alkaptonuria is synonymous to that in the human condition. Osteoarthritis Cartilage. 2012 Aug;20(8):880-6. doi: 10.1016/j.joca.2012.04.013. Epub 2012 Apr 24. |
| [9] | Laschi M, Tinti L, Braconi D, Millucci L, Ghezzi L, Amato L, Selvi E, Spreafico A, Bernardini G, Santucci A. Homogentisate 1,2 dioxygenase is expressed in human osteoarticular cells: implications in alkaptonuria. J Cell Physiol. 2012 Sep;227(9):3254-7. doi: 10.1002/jcp.24018. |
| [10] | Aquaron RR. Alkaptonuria in France: past experience and lessons for the future. J Inherit Metab Dis. 2011 Dec; 34(6):1115-26. doi: 10.1007/s10545-011-9392-7. Epub 2011 Sep 17. Review. |
| [11] | Zatkova A. An update on molecular genetics of Alkaptonuria (AKU). J Inherit Metab Dis. 2011 Dec;34(6):1127-36. doi: 10.1007/s10545-011-9363-z. Epub 2011 Jul 1. |
| [12] | Al-sbou M. Novel mutations in the homogentisate 1,2 dioxygenase gene identified in Jordanian patients with alkaptonuria. Rheumatol Int. 2012 Jun;32(6):1741-6. doi: 10.1007/s00296-011-1868-0. Epub 2011 Mar 25. |
| [13] | Sen Gupta, Parth Sarthi; Bandyopadhyay Amal Kumar. Sequence, Structural and Functional Characterization of Chalcone Synthase of Solanum tuberosum: An in silico analysis; Structural biology(under review) |
| [14] | Castrignanò T, De Meo PD, Cozzetto D, Talamo IG, Tramontano A. (2006). The PMDB Protein Model Database. Nucleic Acids Research, 34: D306-D309. |
| [15] | The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 40: D71-D75 (2012). |
| [16] | Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 1995 Dec;11(6):681-684 |
| [17] | Ishida, T and Kinoshita, K, PrDOS: prediction of disordered protein regions from amino acid sequence., Nucleic Acids Res, 35, Web Server issue, 2007 |
| [18] | Predicting subcellular localization of proteins based on their N-terminal amino acid sequence.Olof Emanuelsson, Henrik Nielsen, Søren Brunak and Gunnar von Heijne.J. Mol. Biol., 300: 1005-1016, 2000. |
| [19] | Arnold K., Bordoli L., Kopp J., and Schwede T. (2006). The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 22,195-201. |
| [20] | James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kale, and Klaus Schulten. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26:1781-1802, 2005. |
| [21] | Lindahl E, Azuara C, Koehl P, Delarue M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. |
| [22] | Debswapna Bhattacharya and Jianlin Cheng. 3Drefine: Consistent Protein Structure Refinement by Optimizing Hydrogen-Bonding Network and Atomic-Level Energy Minimization. Proteins: Structure, Function and Bioinformatics. 2012 (In Press). |
| [23] | GlobPlot: exploring protein sequences for globularity and disorder.Nucleic Acid Res 2003 - Vol. 31, No.13 |
| [24] | Laskowski R A, MacArthur M W, Moss D S, Thornton J M (1993). PROCHECK - a program to check the stereochemical quality of protein structures. J. App. Cryst., 26, 283-291 |
| [25] | Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993 Sep;2(9):1511-9. |
| [26] | Gopalkrishnan K, Sowmiya G, Sheik SS, Sekar K, Ramachandran plot on the web (2.0). PROTEIN PEPT.LETT. (2007), 14,669-671 |
| [27] | Leigh Willard, Anuj Ranjan,Haiyan Zhang,Hassan Monzavi, Robert F. Boyko, Brian D. Sykes, and David S. Wishart "VADAR: a web server for quantitative evaluation of protein structure quality" Nucleic Acids Res. 2003 July 1; 31 (13): 3316.3319 |
| [28] | Mark Berjanskii, Jianjun Zhou, Yongjie Liang, Guohui Lin and David S. Wishart "Resolution-by-Proxy: A Simple Measure for Assessing and Comparing the Overall Quality of NMR Protein Structures", J Biomol NMR. 2012 Jul;53(3): 167-80 |
| [29] | Zengming Zhang, Yu Li, Biaoyang Lin, Michael Schroeder and Bingding Huang (2011), Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics, 27 (15): 2083-2088. |
| [30] | Sigrist CJA, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, Bougueleret L, Xenarios I. New and continuing developments at PROSITE Nucleic Acids Res. 2012; doi: 10.1093/nar/gks1067 |
| [31] | (My Hits, SIB, Switzerland) includes Prosite, Pfam and HAMAP profiles. |
| [32] | A Hierarchical Approach to Protein Fold Prediction Tabrez Anwar Shamim Mohammad1 and Hampapathalu Adimurthy Nagarajaram2 Laboratory of Computational Biology, CDFD, Bldg.7, Gruhakalpa, Nampally, Hyderabad |