-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2013; 3(2): 30-33
doi:10.5923/j.bioinformatics.20130302.04
PDB-by-RMSD: The New Service and Tool for Searching Protein Structures
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLA. S. Shevnin, A. D. Semenov, Yu. B. Porozov
Laboratory of bioinformatics, Saint-Petersburg National Research University of Information Technologies, Mechanics and Optics, Kronverkskiy pr. 49, Saint-Petersburg, 197101, Russian Federation
Correspondence to: Yu. B. Porozov, Laboratory of bioinformatics, Saint-Petersburg National Research University of Information Technologies, Mechanics and Optics, Kronverkskiy pr. 49, Saint-Petersburg, 197101, Russian Federation.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Modern databases and search engines for structural biology are very sophisticated and versatile. However, scientific life discovers new research topics almost every day. For instance scientific visualization is a field where distances between different conformations play a crucial role. In our research of protein motions we have faced a problem of searching of structures with known Root Mean Square Deviation (RMSD). In spite of the fact that there is a set of tools giving possibility of search on different databases, it is impossible to set RMSD interval between NMR models of some protein as the search criterion. PDB-by-RMSD is a tool, developed to provide such possibility. It can be used in protein motion researching or protein visualization[1–5].
Keywords: Protein Motion, RMSD, Conformation, Scientific Visualization, Geometry Modeling
Cite this paper: A. S. Shevnin, A. D. Semenov, Yu. B. Porozov, PDB-by-RMSD: The New Service and Tool for Searching Protein Structures, American Journal of Bioinformatics Research, Vol. 3 No. 2, 2013, pp. 30-33. doi: 10.5923/j.bioinformatics.20130302.04.
Article Outline
1. Introduction
- Now, most of internet protein data storages provides some search tool. Typical parameters are Identifier, Pubmed Id, Description, Keywords, Release Date, some numerical values. At the same time, one cannot search by calculation-depending parameters, such as Root Mean Square Deviation between different conformations of one structure. This value, while is not optimal measure of structural differences in some cases, still is very important in almost all fields of structural biology of proteins. For instance in[5] authors tried to compose a path through multiple conformations of Apo-Calmodulin on the basis of RMSD ordering. So, we had to develop service for easy searching structures by this parameter. Distance between conformations serves as crucial factor for modelling technique choice when it is necessary to build a trajectory of protein’s behaviour. Sometimes it is possible to use various local tools for RMSD calculation (for instance Trajectory Tool in VMD package[6]). But these tools are not able to select structures from PDB.org with RMSD required. The only online service and database that provide a kind of search by RMSD is Protein Conformation Database, PCDB[7]. Although PCDB provides some kind of RMSD selection in our opinion our tool offer more quick, clear and straight search directly in PDB database.PDB-by-RMSD is a tool that provides a simple and easy-to-use interface for searching of protein structures in the PDB archive[8] by their RMSD. Search can be performed by several parameters but the main purpose of the tool is to provide structures selected by RMSD range specified by users.The aim of the paper is to describe the new search tool PDB-by-RMSD. We describe the core organization of this tool and its functionality. Then we explain fields of the form for requests and show how PDB-by RMSD solves user’s requests.
2. Materials and Methods
- PDB-by-RMSD has been written using the C# programming language and .NET Framework platform. Following libraries has been used: Microsoft .NET Framework 4.0, Math.NET Numerics, zlib.net and Google protobuf. Source code and binaries are available on demand.The PDB-by-RMSD application supports work with the PDB.org archive and its copy on a local machine.The source PDB archive located on the PDB.org server takes about 160 Gb, so one-by-one entities processing could take a lot of time (because PDB service doesn’t support RMSD value as query parameter and each entity has to be downloaded on local machine to be processed). Therefore a local database was developed. It stores all PDB files in a compressed format and contains all required information of the original database such as keywords, atom coordinates, etc. Due to the local databases structure the fully updated local database takes about 50 Gb.The local database supports synchronization with PDB.org and other databases. This functionality is provided by plugins.Basically, any external database engine could be used as database (for instance, organization’s SQL server). Core library contains set of public .NET interfaces (CommonLib. Querying. IDataLoader and CommonLib. Querying. Idata Saver), which has to be implemented and compiled as dynamic libraries to provide such functionality. One doesn’t need to recompile core libraries or use its source code to add new data providers.Current implementation of local database keeps structures in binary format. Besides, all information that could be used for fast search (Id, keywords, RMSD values) is in one small index file.The PDB archive updates on Wednesdays so you should update your local database on these days to use actual data.PDB-by-RMSD service interacts with Protein Data Bank using its SOAP service. Plugin system was developed due to flexibility purposes. Plugins can add new RMSD minimisation methods or new data source providers. Local data base is also controlled by plugin, so it can be replaced by any relational database.We use a simple and fast algorithm of structure alignment to avoid possible solid-state transitions and rotations. The web-service minimizes RMSD by the method based on three point superimposition and quaternions[9]. Then PDB-by- RMSD calculates various types of RMSD, such as full-atomic distances with or without hydrogen, Cα trace RMSD, main chain RMSD with or without oxygen. These values are indexed and stored in local tables for very fast solving requests and reporting.
3. Results and Discussion
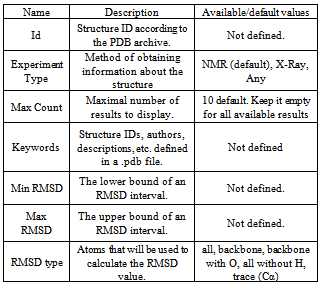
- The developed tool is available in two types as web-service and as standalone application.The web-service is a part of our laboratory site and available at http:// bioinfolab. ifmo. ru/ Services/ Show Service RMSD. It provides an ability to search structures in the PDB archive by the following parameters: RMSD, Structure ID, Expirement Type, Keywords. The standalone application is also available.By clicking on the structures ID you will be redirected to the PDB.org page containing detailed information about this structure. The RMSD value is calculated only for structures whose information was obtained by the NMR Experiment Type.All requests and their results are cached, so you can get results for common requests fast enough. Having pressed the Calculate button, you will be redirected to the URL which uniquely defines your request. Opening this URL on any computer the same page with cached request results will be displayed. It can be useful for complicated requests that can be processed for a long time because you don’t need to keep your browser open. Remember the requests URL and repeat it later to see your results.The table 1 contains descriptions of the search parameters, their available and default values.
|
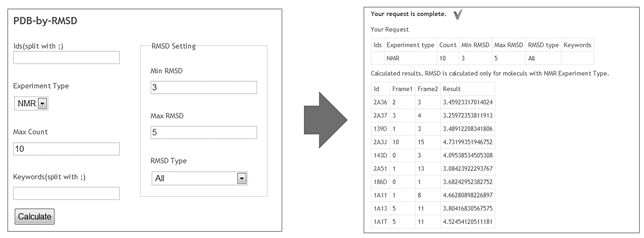 | Figure 1. Left form contains example of search request. In this case user is going to find 10 entities with RMSD from 3 to 5. Query results are on the right pane |
4. Conclusions
- It is obvious today that not only structure determines function of proteins but also their behaviour[10]. This consideration results in development of numerous techniques for protein motions modelling. Methods and modifications of molecular dynamics (MD) are one of well known and well developed. At the same time MD still has numerous of limitations of implementation – time scale, number of atoms, local energy barriers and calculation expenses. In order to override these limitations lot of coarse-graned models were developed[11-14] as well as field of so called scientific visualization and animation. Both MD approaches, coarse-graned and animation approaches are very sensitive to distances between conformations of the protein of interest. Even choice of a method to apply is related with RMSD. The quality of results obtained is under in under influence of structural differences. Another motivation of this work is absence of RMSD information in .pdb records and impossibility to run such searches easily.In this work we introduced the new service for fast search of proteins by RMSD between conformations. By applying a local database and indexing search requests are processed very quickly. Many types of RMSD are available for user as well as keyword filtration. In many fields of structural modelling from molecular dynamics to scientific animation the distance between models is very important for method choosing and analysis of results. This new instrument for PDB search by RMSD criterion is available for scientific community.
ACKNOWLEDGMENTS
- We are grateful to Anton Neyolov for reading this paper and for advices.This work was supported by the Ministry of Education and Science of Russian Federation (federal special purpose program of Russian Federation "Research and development on priority directions of scientific-technological complex of Russia for 2007-2013" № 14.514.11.4068).