-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2012; 2(4): 61-67
doi:10.5923/j.bioinformatics.20120204.05
Comparison Between Artificial Immune System and other Heuristic Algorithms for Protein Structure Prediction
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLR. F. Mansour1, Faraj Al-Ghamdi2
1Department of Science and Mathematics , Faculty of Education, New Valley,Assiut University 71516, EL kharaga, Egypt
2Department of Biology Science, Faculty of Science, Northern Borders University, KSA
Correspondence to: R. F. Mansour, Department of Science and Mathematics , Faculty of Education, New Valley,Assiut University 71516, EL kharaga, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The search for the global minimum of a potential energy function is very difficult since the number of local minima grows exponentially with the molecule size. We present an Artificial Immune System (AIS) inspired by the clonal selection principle, which has been designed for the protein structure prediction problem (PSP). The proposed AIS employs two special mutation operators, hypermutation and hypermacromutation to allow effective searching, and an aging mechanism which is a new immune inspired operator that is devised to enforce diversity in the population during evolution. When cast as an optimization problem, the PSP can be seen as discovering a protein conformation with minimal energy. Our experimental results demonstrate that the proposed AISis very competitive with the existing state-of-art algorithms for the PSP on lattice models with low computational costs.
Keywords: Immune System, Heuristic Algorithm, Genetic Algorithm, Protein Structure Prediction
Cite this paper: R. F. Mansour, Faraj Al-Ghamdi, Comparison Between Artificial Immune System and other Heuristic Algorithms for Protein Structure Prediction, American Journal of Bioinformatics Research, Vol. 2 No. 4, 2012, pp. 61-67. doi: 10.5923/j.bioinformatics.20120204.05.
Article Outline
1. Introduction
- Artificial Immune Systems (AISs) represent a field of biologically inspired computing that attempts to exploit theories, principles, and concepts of modern immunology to design immune system-based applications in science and engineering[1–3]. The IS provides an excellent example of a bottom up intelligent strategy[4], through which adaptation operates at the local level of cells and molecules, and useful behavior emerges at the global level: this is exemplified by the immune humeral and cellular responses.AISs are proving to be a very general and applicable form of bio-inspired computing. A great deal of work has gone into developing algorithms that extrapolate basic immune processes such as clonal selection, negative and positive selection, danger theory, and immune networks[2]. To date, AIS have been applied to areas such as machine learning[5],[6], optimization[7],[8], bioinformatics[9 –11], robotic systems[12–14], decision support systems[15], network intrusion detection[16],[17], combinatorial optimization[18],[19] and scheduling[20], Given the primary sequence of a protein, the protein structure prediction problem requires the identification of its native (tertiary) conformation with minimum energy; while the protein folding problem requires information about the possible pathways to folding and unfolding. Since a protein’s structure determines its biological function, it is very important to be able to predict the final spatial conformation of the proteins. In this work, the standard HP lattice bead model has been incorporated into an immune algorithm. Despite the minimalistic approach employed by this model, it has been shown to belong to the “NP-Hard” set of problems, and many approximation methods and heuristics have been investigated to address it[21]. Existing computing approaches to PSP include evolutionary search[22-25], ant colony optimization[26], memetic algorithms[27], tabu search[28] and Monte Carlo algorithms[29].The potential energy of a molecule is derived from molecular mechanics, whichdescribes molecular interactions based on the principles of Newtonian physics. Anempirically derived set of potential energy contributions is used for approximatingthese molecular interactions.This set of potential energy contributions, called theforce field, contains adjustableparameters that are selected in such a way as toprovide the best possible agreementwith experimental data. Our discussion will focuson energy functions which sharethe main features of general molecular force fields.The molecular model considered here consists of a chain of m atoms centered at
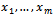 in a 3D space, as seen in Figure 1. For every pair of consecutive atoms xi and
in a 3D space, as seen in Figure 1. For every pair of consecutive atoms xi and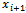 , let
, let  be the bond length which is the Euclidean distance between them. For every three consecutive atoms xi, ,
be the bond length which is the Euclidean distance between them. For every three consecutive atoms xi, , 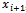
 let
let be the bond angle corresponding to the relative position of the third atom with respect to the line containing the previous two. Likewise, for every four consecutive atoms
be the bond angle corresponding to the relative position of the third atom with respect to the line containing the previous two. Likewise, for every four consecutive atoms 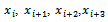 let
let 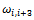 be the angle, called the torsion angle, between the normal through the planes determined by the atoms
be the angle, called the torsion angle, between the normal through the planes determined by the atoms and
and  The force field potentials corresponding to bond lengths, bond angles, and torsion angles are defined respectively[30] as
The force field potentials corresponding to bond lengths, bond angles, and torsion angles are defined respectively[30] as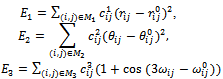 | (1) |
 is the bond stretching force constant,
is the bond stretching force constant, 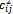 is the angle bending force constant, and
is the angle bending force constant, and is the torsion force constant. The constant
is the torsion force constant. The constant 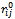 and
and 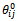 represent the “preferred” bond length and bond angle, respectively, and the constant
represent the “preferred” bond length and bond angle, respectively, and the constant 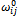 is the phase angle that defines the position of the minima. The set of pairs of atoms separated by k covalent bond is denoted by
is the phase angle that defines the position of the minima. The set of pairs of atoms separated by k covalent bond is denoted by for k = 1, 2, 3. In addition to the above, there is a potential
for k = 1, 2, 3. In addition to the above, there is a potential 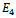 which characterizes the 2-body interaction between every pair of atoms separated by more than two covalent bonds along the chain. We use the following function to represent
which characterizes the 2-body interaction between every pair of atoms separated by more than two covalent bonds along the chain. We use the following function to represent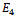
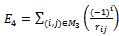 | (2) |
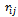 is the Euclidean distance between atoms
is the Euclidean distance between atoms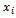 and
and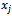 .The general problem is the minimization of the total molecular potential energy function,
.The general problem is the minimization of the total molecular potential energy function,  , leading to the optimal spatial positions of the atoms. To reduce the number of parameters involved in the potentials above, we simplify the problem considering a chain of carbon atoms. In this case, it is known that the preferred bond lengths are
, leading to the optimal spatial positions of the atoms. To reduce the number of parameters involved in the potentials above, we simplify the problem considering a chain of carbon atoms. In this case, it is known that the preferred bond lengths are = 1.526°A (for all
= 1.526°A (for all  and that the bond angles are
and that the bond angles are  for all
for all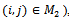 , we consider also that
, we consider also that 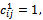 for all
for all  for all
for all 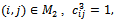 for all
for all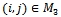 and
and  for all
for all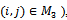 , While this structure reflects great simplifications over the general problem, its complexity should not be underestimated, as we will see below. In most molecular conformational predictions, all covalent bond lengths and covalent bond angles are assumed to be fixed at their equilibrium values
, While this structure reflects great simplifications over the general problem, its complexity should not be underestimated, as we will see below. In most molecular conformational predictions, all covalent bond lengths and covalent bond angles are assumed to be fixed at their equilibrium values  and
and  respectively. Thus, the molecular potential energy function reduces to
respectively. Thus, the molecular potential energy function reduces to 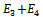 and the first three atoms in the chain can be fixed. The first atom,x1 ,is fixed at the origin, (0, 0, 0); the second atom,x2 , is positioned at
and the first three atoms in the chain can be fixed. The first atom,x1 ,is fixed at the origin, (0, 0, 0); the second atom,x2 , is positioned at 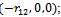 and the third atom, x3, is fixed at
and the third atom, x3, is fixed at 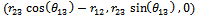 Using the parameters previously defined and Eq.(1) and Eq.(2), we obtain
Using the parameters previously defined and Eq.(1) and Eq.(2), we obtain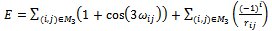 | (3) |
 , Eq.(1), is expressed as a function of torsion angles, and E4 , Eq.(2), is expressed as a function of Euclidean distance. To represent Eq.(3) as a function angles only, we can use the result established in[32] and obtain
, Eq.(1), is expressed as a function of torsion angles, and E4 , Eq.(2), is expressed as a function of Euclidean distance. To represent Eq.(3) as a function angles only, we can use the result established in[32] and obtain | (4) |
 | (5) |
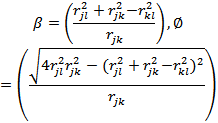 and
and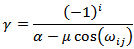 where i= 1, . . . ,m − 3 and m is the number of atoms in the given system as seenin Figure 1.Despite these simplification, the problem remains very difficult. A molecule with as few as 30 atoms has 227 = 134, 217, 728 local minimizers. It can clearly beseen that finding the global minimum for chains of even moderate length is intractable via exhaustive methods[33].
where i= 1, . . . ,m − 3 and m is the number of atoms in the given system as seenin Figure 1.Despite these simplification, the problem remains very difficult. A molecule with as few as 30 atoms has 227 = 134, 217, 728 local minimizers. It can clearly beseen that finding the global minimum for chains of even moderate length is intractable via exhaustive methods[33]. | Figure 1. Coordinate Set of Atomic Chain |
2. Llattice model HP for the PSP
- In this work, the standard HP lattice bead model is embedded in a 2-dimensional square lattice, restricting bond angles to only a few discrete values. Interactions are only counted between topological neighbours, that is between beads (representing amino acids) that lie adjacent to each other on the lattice, but which are not sequence neighbours . The energies corresponding to the possible topological interactions are as follows:
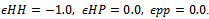 By summing over these local interactions, the energy of the model protein can be obtained:
By summing over these local interactions, the energy of the model protein can be obtained: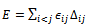 | (6) |
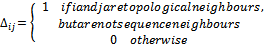 The HP lattice model recognizes only the hydrophobic interaction as the driving force in protein folding, with many native structures protecting the hydrophobic core with polar residues, resulting in a compact arrangement[34].
The HP lattice model recognizes only the hydrophobic interaction as the driving force in protein folding, with many native structures protecting the hydrophobic core with polar residues, resulting in a compact arrangement[34].
|

3. The Immune Algorithm
- An immune algorithm[36] is inspired by the clonal selection principle employed by the human immune system. In this process, when an antigen enters the body, B and T lymphocytes are able to clone upon recognition and bind to it[36]. Many clones are produced in response and undergo many rounds of somatic hypermutation. The higher the fitness of a B cell to the available antigens, the greater the chance of cloning. Cells have a certain life expectancy, allowing a higher specific responsiveness for future antigenic attack[34] .The IA presented here includes the aging, cloning and selection operators used in a previous study by Cutello et al.[34], with modified constructor and mutation operators. The constructor employs a backtracking algorithm that records some of the possible mutations by testing bead placement during chain growth. These possibilities are exploited and updated during the mutation process, preventing an infeasible conformation from occurring based on the preceding self-avoiding structure for a particular point in the model protein chain. In retaining this information, infeasible mutations are not explored, allowing a greater number of constructive mutations to be investigated. Figure 2 illustrates the stages involved in placing two consecutive beads during the chain growth phase. Before committing a bead to the lattice, all possible directions are explored, Figure 2(a), and from the valid options available, a random choice is made, Figure2(b). Again all possible choices are investigated, marking any infeasible options (note that choosing left will not result in a self avoiding conformation), Figure2(c), and a valid choice is selected from the remaining options, Figure 2(d). Any remaining valid choices are left unmarked for use in the first mutation phase after the initial chain growth. Once a valid mutation has been made, the entire structure is reconstructed as before marking any infeasible directions as a result of the new conformation vector.In the basic IA set up, there are a maximum of 1,000,000 fitness evaluations, with the maximum number of generations set to 50,000. In order to estimate the optimal combination of parameters, we adopted the procedure used by Cutelloet al., whereby the maximum Bcell age and the number of clones were each varied from 1 to 10. Population sizes examined were 10, 25, 50, 100 and 200. This provided a combination of 500 different parameter sets for each sequence, which was applied to allthe benchmark sequences up to 25 beads in length. All fitness evaluations for the best success rates were collated and graded for overall performance. As a result of this preliminary testing, the results presented below were obtained using a maximum B-Cell age of 4, 3 clones and a popu
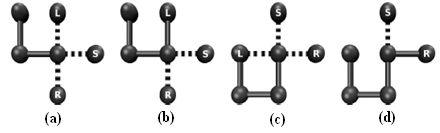 | Figure 2. Stages of the chain growth algorithm investigating left (L), right (R) and straight (S) availability (a), random selection from all available directions (b), at the next locus investigation of L, R and S availability (c) and random selection from remaining available S and R directions (d) |
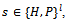 where lis the protein length, i.e., the number of amino acid in the protein sequence, whereThe B cell population,
where lis the protein length, i.e., the number of amino acid in the protein sequence, whereThe B cell population, 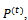 represents a set of candidate solutions in the current fitness landscape at each generation t. The B cell, or B cell receptor, is a sequence of directions
represents a set of candidate solutions in the current fitness landscape at each generation t. The B cell, or B cell receptor, is a sequence of directions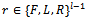 (with L=Left, R=Right and F= Forward), where eachri , with
(with L=Left, R=Right and F= Forward), where eachri , with  is a relative direction with respect to the previous direction
is a relative direction with respect to the previous direction (i.e., there are
(i.e., there are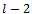 relative directions) and
relative directions) and 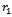 the nonrelative direction.Hence, we obtain an overall sequence of length
the nonrelative direction.Hence, we obtain an overall sequence of length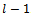 . The sequence r specifies a 2-D conformation which is suitable for computing the energy value of the hydrophobic-pattern of the given protein. At each generation t , there is a B cell population of
. The sequence r specifies a 2-D conformation which is suitable for computing the energy value of the hydrophobic-pattern of the given protein. At each generation t , there is a B cell population of 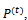 size d. The initial population, time t=0, is randomly generated in such a way that each B cell of
size d. The initial population, time t=0, is randomly generated in such a way that each B cell of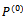 , is a self-avoiding conformation. There are two main functions within the algorithm. Evaluate(P) which computes the fitness function value of each B cell
, is a self-avoiding conformation. There are two main functions within the algorithm. Evaluate(P) which computes the fitness function value of each B cell ; hence
; hence 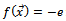 is the energy of conformation coded in the B cell receptor
is the energy of conformation coded in the B cell receptor ; and Termination_Condition()which returns true if a solution is found, or a maximum number of fitness function evaluations
; and Termination_Condition()which returns true if a solution is found, or a maximum number of fitness function evaluations 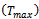 is reached. The implemented IA, like all IAs based on the clonal selectionprinciple outlined above, is characterized by cloning of B cells with higher antigenic affinity, affinity maturation, and hypermutation of offspring B cells.
is reached. The implemented IA, like all IAs based on the clonal selectionprinciple outlined above, is characterized by cloning of B cells with higher antigenic affinity, affinity maturation, and hypermutation of offspring B cells. 4 Results
4.1. Algorithm Comparison
- With CPU time being hardware dependent, the number of fitness evaluations (together with the percentage success rate) has been used to assess the efficiency of the algorithm, as shown in Table 2 for the benchmark sequences. It is apparent from Table 2 that, although the use of memory B-Cells[34] hinders the discovery of global minima for some of the smaller sequences, it enhances the search forthe larger, more difficult to find sequences. The memory ability allows mid to high fitness conformations to remain in the population for a longer number of generations. For larger sequences, this allows a more detailed exploration in certain areas of the potential energy surface, permitting thememory B-Cells to converge towards the global solution much sooner. In contrast, for smaller sequences the mid to high fitness range is much smaller, thereby preventing a rapid exploration of the potential energy surface by retaining unfavorable segments of local structure for a largernumber of generations. Generally, the use of memory Bcells allows a more diverse inspection of the potential energy surface, due to a greater number of the degenerate conformations being found. This is achieved as favourable fragments of local structure are not rapidly disposed of during the retirement process, hindering efficiency as a consequence. The algorithm presented here shows promising results,being comparable to the work of Cutelloet al.[34]. While our success rates for the larger sequences (e.g. HP-48) are a little lower, in some cases our numbers of fitness evaluations show an improvement.The compact structural arrangement present in all globalminima (GMs) is apparent from the example GMs shown in Figure3. With the driving force being the hydrophobic topological contact, it can be seen that compact hydrophobiccores give rise to high fitness conformations. Inspection of the HP-48 global minimum (i) allows us to understand the poor success rate for this sequence. The 5x5 hydrophobic core presents a problem to the IA in achieving convergence, as a single misplaced hydrophobic bead will result in only a metastable conformation. The problem does not exist for the HP-50 sequence (j), due to the presence of two small hydrophobic cores coupled by a chain of hydrophobic beads, which explains the higher success rate and fewer average structure evaluations necessary for HP-50, compared with HP- 48 and (when using memory B-cells) even the much shorter HP-36 sequence . The work of Cutello et al. supports this idea[34], as similar magnitudes of the number of fitness evaluations for these problematic sequences can be seen, with a much lower success rate for HP-48 than for any other instance
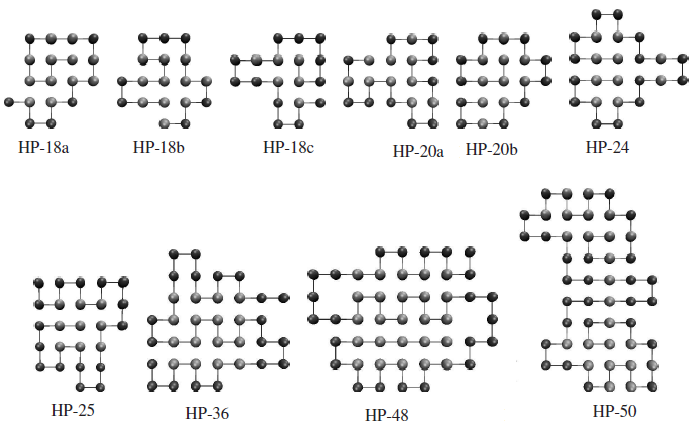 | Figure 3. Examples of GM structures for the benchmark sequences |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.2. Comparisons with Other Evolutionary Algorithms
- The results of the proposed algorithm for the HP instances are directly compared to the best results obtained by the following related evolutionary models: the results of standard GA[36], Pull-Move GA (PMGA)[37], multimeme algorithms (MMA)[27], and estimation of distribution algorithms (EDAs)[38]. The results of GA[36] are based on a population of 200 structures evolved over 300 generationsTable 3 presents the known optimum for each protein instance and the energy values found by each of the compared methods. A direct comparison between energy values obtained by the different evolutionary models emphasizes a good and competitive performance of the proposed ELF method. The results are clearly better than those reported by GAs[36] and PMGAs[37]. PMGA[37] is included in the comparative analysis as it is an evolutionary algorithm using pull move transformations in addition to the standard genetic operators (pull moves are not applied in a hill-climbing mode like in the IA and a specialized fitness function is used). PMGA improves the results of standard GAs for PSP but reports worse results for sequences S5, S7 and S8 compared to IA. MMAs represent an interesting approach to be compared with IA as both models use local search but according to different strategies. Results indicate a better solution obtained by IA for instance S5 when compared to MMA. This is a promising result for the proposed method underlying the good performance of hill-climbing crossover and pull-moves. Furthermore, the results of IA are competitive with those reported by recently proposed algorithm EDA. For sequence S7, IA obtains same global best energy of −35 while for sequence S8, IA is able to identify a higher energy conformation compared to other but does not detect the known optimum energy configuration as EDA.
5. Conclusions
- Although implementation of a modified constructor for usein the mutation phase of the IA has not always given greatersuccess rates (especially for more challenging sequences),it has allowed for a more efficient search to be performedin some cases, showing a decrease in the number of fitnessevaluation performed. The use of population diversitytracking allows a greater understanding of the algorithm’sability to explore areas of the potential energy surface ofthese simple model proteins. Areas of favourable localstructure along the chain can be assessed, illustrating theimportant allele combinations that give rise to the determinationof global minima. We are currently applying theseapproaches to more realistic protein models
References
| [1] | L.N. de Castro and J. Timmis.Artifical Immune Systems and Their Applications. Springer-Verlag, 1999 |
| [2] | L. N. de Castro and J. Timmis, Artificial Immune Systems: A NewComputationalIntelligence Paradigm. London, U.K.: Springer-Verlag,2002. |
| [3] | Y. Ishida, Immunity-Based Systems: A Design Perspective.eidelberg, Germany: Springer-Verlag, 2004. |
| [4] | V. Cutello and G. Nicosia, “The clonal selection principle for in silicoand in vitro computing,” in Recent Developments in Biologically Inspired computing, L.N. de Castro and F. J. von Zuben, Eds. Hershey,PA: Idea Group Publishing, 2004. |
| [5] | J. E. Hunt and D. E. Cooke, “Learning using an artificial immunesystem,” J. Netw. Comput.Appl., vol. 19, pp. 189–212, 1996. |
| [6] | G. Nicosia, F. Castiglione, and S. Motta, “Pattern recognition by primaryand secondary response of an artificial immune system,” Theoryin Biosciences, vol. 120, no. 2, pp. 93–106, 2001. |
| [7] | T. Fukuda and M. T. K. Mori, “Parallel search for multimodal functionoptimization with diversity and learning of immune algorithm,” inArtif.Immune Syst. Their Appl., D. Dasgupta, Ed. Berlin, Germany:Springer-Verlag, 1999. |
| [8] | L. N. de Castro and F. J. V. Zuben, “Learning and optimization usingthe clonal selection principle,” IEEE Trans. Evol. Comput., vol. 6, pp.239–251, Jun. 2002. |
| [9] | G. Nicosia, “Immune algorithms for optimization and protein structureprediction,” Ph.D. dissertation, Dept. Math. Comput.Sci., Univ.Catania, Catania, Italy, 2004. |
| [10] | G. B. Bezerra, L. N. de Castro, and F. J. V. Zuben, “A hierachicalimmune network applied to gene expression data,” in Proc. 3rd Int.Conf. Artif. Immune Syst., G. Nicosia, V. Cutello, P. Bentley, and J.Timmis, Eds., Catania, Italy, pp. 14–27,2004. |
| [11] | V. Cutello, G. Narzisi, and G. Nicosia, “A multi-objective evolutionaryapproach to the protein structure prediction problem,” J. Royal So. Interface,vol. 3, no. 6, pp. 139–151, Feb. 2006. |
| [12] | S. Ichikawa,A. Ishiguro, S.Kuboshiki, and Y. Uchikawa, “Amethod ofgait coordination of hexapod robots using immune networks,” J. Artif.Life Robotics, vol. 2, pp. 19–23, 1998. |
| [13] | S. Singh and S. Thayer, “Kilorobot search and rescue using animmunologicallyinspired approach,” in Distributed Autonomous Robotic Systems.Berlin, Germany: Springer-Verlag, , vol. 5,2002. |
| [14] | L. Kesheng, Z. Jun, C. Xianbin, and W. Xufa, “An algorithm basedon immune principle adopted in controlling behavior of autonomousmobile robots,” Comput. Eng. Appl., vol. 5, pp. 30–32, 2000. |
| [15] | D. Dasgupta, “An artificial immune system as a multiagent decisionsupport system,” in Proc. IEEE Int. Conf. Syst., Man, Cybern., SanDiego, CA, , pp. 3816–3820,1998. |
| [16] | J. Kim and P. Bentley, “Towards an artificial immune system for networkintrusion detection: An investigation of clonal selection with negativeselection operator,” in Proc. IEEE Int. Congr. Evol.Comput.,Seoul, Korea, pp. 1244–1252, 2001. |
| [17] | D. Dasgupta and F. A. Gonzalez, “An immunity-based techniqueto characterize intrusions in computer networks,” IEEE Trans. Evol.Comput., vol. 6, pp. 281–291, 2002. |
| [18] | V. Cutello and G. Nicosia, “An immunological approach to combinatorialoptimization problems,” in Proc. 8th Ibero-American Conf. Artif.Intell., Seville, Spain, , pp. 361–370, 2002. |
| [19] | V. Cutello, G. Nicosia, and M. Pavone, “A hybrid immune algorithmwith information gain for the graph coloring problem,” in Proc. LNCSon Genetic and Evol.Comput. Conf., Chicago, IL, vol. 2723,pp. 171–182, 2003. |
| [20] | E. Hart and P. Ross, “The evolution and analysis of a potential antibodylibrary for use in job-shop scheduling,” in New Ideas in Optimization,D. Corne, M. Dorigo, and F. Glover, Eds. London, U.K.: McGraw-Hill, 1999. |
| [21] | Zhao, X.: Advances on protein folding simulations based on the latticeHP models with natural computing. Appl. Soft Comput., 8, 2, 1029–1040,2008. |
| [22] | Berenboym, I., Avigal, M.: Genetic algorithms with local search optimization for protein structure prediction problem. GECCO 2008, 1097–1098, 2008. |
| [23] | Cotta, C.: Protein Structure Prediction Using Evolutionary Algorithms Hybridized with Backtracking. Artificial Neural Nets Problem Solving Methods, 7th International Work-Conference on Artificial and Natural Neural Networks, LNCS 2687, 321–328, 2003. |
| [24] | Song, J., Cheng, J., Zheng, T., Mao, J.: A Novel Genetic Algorithm for HP Model Protein Folding. PDCAT ’05: Proceedings of the Sixth International onference on Parallel and Distributed Computing Applications and Technologies, IEEE Computer Society, 935–937, 2005. |
| [25] | Unger, R., Moult, J.: Genetic algorithms for protein folding simulations. J. Molec. Biol., 231, 75–81, 1993. |
| [26] | Shmygelska, A., Hernandez, R., Hoos, H.H.: An Ant Colony Algorithm for the 2D HP Protein Folding Problem. In Proc. of ANTS 2002, LNCS, 2463,p. 40-53, 2002. |
| [27] | Krasnogor, N., Blackburnem, B., Hirst, J.D., Burke, E.K.: Multimeme algorithms for protein structure prediction. In 7th International Conference Parallel Problem Solving from Nature (PPSN), LNCS 2439, 769–778, 2002. |
| [28] | Lesh, N., Mitzenmacher, M., Whitesides, S.: A complete and effective move set for simplified protein folding. RECOMB ’03: Proceedings of the seventh annual international conference on Research in computational molecular biology, ACM, 188–195, 2003. |
| [29] | Hsu, H.P., Mehra, V., Nadler, W., Grassberger, P.: Growth algorithms for lattice heteropolymers at low temperatures, J. Chem. Phys., 118, 1, 444– 451, 2003. |
| [30] | M. Dra˘zi´c, C. Lavor, N. Maculan and N. Mladenovi´c, A continuousvariable neighborhoodsearch heuristic for finding the three-dimensional structure of a molecule, European Journalof Operational Research, 185 , 1265–1273, 2008. |
| [31] | D. J. Wales, H. A. Scheraga, Global optimization of clusters, crystals and biomolecules,Science, 285 , 1368–1372, 1999 |
| [32] | A.Hedar, A.F. and T. Hassan ,"Gentic Algorithm and Tabu Searched Methods for Molecular 3D-Structure Predicition", Numerical Algebra Control and Optimization, Vol. 1, No. 1, pp. 191–209, 2011 |
| [33] | C. Lavor and N. Maculan, A function to test methods applied to global minimization of potential energy of molecules, Numerical Algorithms, 35 , 287–300, 2004. |
| [34] | V. Cutello, G. Niscosia, M. Pavone, and J. Timmis.An Immune Algorithm for Protein Structure Predictionon Lattice Models. IEEE T. Evol. Comput.,11(1), pp.101–117, 2007. |
| [35] | W.E. Hart, S. Istrail. HP Benchmarks ww.cs.sandia.gov/tech_reports/compbio/tortilla-hp-benchmarks.html. |
| [36] | Unger, R., Moult, J.: Genetic algorithms for protein folding simulations. J. Molec. Biol., 231, 75–81, 1993. |
| [37] | Berenboym, I., Avigal, M.: Genetic algorithms with local search optimization for protein structure prediction problem. GECCO 2008, 1097–1098, 2008. |
| [38] | R. Santana, P. Larra˜naga, and J. A. Lozano. Protein folding in simplified models with estimation of distribution algorithms. IEEE Transactions on Evolutionary Computation.Vol. 12.No. 4.pp. 418-438, 2008. |