-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
p-ISSN: 2167-6992 e-ISSN: 2167-6976
2012; 2(4): 55-60
doi: 10.5923/j.bioinformatics.20120204.04
Structural and Functional Interferences from a Molecular Structural Model of Xenocin Toxin from Xenorhabdus nematophila
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLJitendra Singh
School of Biotechnology, Gautam Buddha University, Greater Noida, 201308 Uttar Pradesh, India
Correspondence to: Jitendra Singh , School of Biotechnology, Gautam Buddha University, Greater Noida, 201308 Uttar Pradesh, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Bacteriocins constitute the most abundant and diverse family of microbial defense systems. They are produced and secreted to inhibit the growth of closely related, competing bacterial species inhabiting a common ecological niche. One such bacteriocin designated as xenocin, is produced by entomopathogenic bacterium Xenorhabdus nematophila. In our earlier study, we have shown its regulation under SOS conditions and its activity against wide range of bacteria isolated from the gut of insect. In this study three dimensional structure of xenocin has been deciphered with the assistance of automated homology modelling and verified by VERIFY-3D program as well as Ramachandran plot 2.0. Three domain organisation; Translocation (T), Receptor (R) and Catalytic domain (C) has been observed and their structures were studied by CHIMERA software (UCSF). Protein disorder structure and Average Area Buried Upon folding (AABUF) in first 100 amino acid residues of Translocation domain is determined by Globplot and ProtScale software respectively. Conserved amino acid residues in the putative active site of the catalytic domain of xenocin have been deciphered with multiple sequence alignment.
Keywords: X. Nematophila, Xenocin, Homology Modeling, Translocation, Receptor , Catalytic Domain
Article Outline
1. Introduction
- Bacteriocins are ribosomally encoded toxins produced by bacteria to inhibit the growth of closely related bacterial strain(s)[1]. They are produced by almost all the major lineages of Eubacteria and Archebacteria and are structurally, functionally, ecologically diverse[2]. Bacteriocins are generally produced in the cytoplasm and secreted in the extracellular medium by the producer where they target specific receptors on the surface of susceptible cells. Induction of toxicity in target cells occurs by different mechanisms including enzymatic nuclease (DNase or RNase) as well as pore formation in the cytoplasmic membrane[3]. Their structure comprises of three distinct domains organization: (i) a domain involved in recognition of specific receptor, (ii) a domain involved in translocation, and (iii) a domain responsible for their toxic activity, with average molecular mass of ~25 to 80 kDa[4].Xenorhabdus nematophila is a motile gram-negative entomopathogenic bacterium belonging to the family Enterobacteriaceae[5]. It forms symbiotic association in the gut of a soil nematode of family Steinernematidae[6]. X.nematophila is known to secrete several extracellular products, which include lipase(s), phospholipase (s), protease(s), and several broad spectrum antibiotics[7,8] when grown under standard laboratory conditions. These products are believed to be secreted in the insect hemolymph during the stationary phase of bacterial growth. The degradative enzymes are responsible for the breakdown of macromolecules of the insect cadaver to provide nutrient to the developing nematode, while the antibiotics play a major role in the suppression of contamination of the cadaver by other soil microorganisms. In our earlier study, we have isolated and characterized xenocin operon encoded by the genome of X. nematophila. Results showed that the transcription of xenocin was upregulated by iron depleted condition, high temperature and in the presence of mitomycin C. Recombinant xenocin-immunty protein complex showed broad range of antibacterial effect, not only limited to the laboratory strains but also to the bacteria isolated from the gut of H. armigera [9]. Therefore, it is essential to study the structure of such an important antibacterial protein in detail. In this study, similar proteins from other sources has been identified with protein-protein blast and phylogenetically analyzed. Three dimensional structiure of xenocin is deciphered by automated homology modeling. Protein disorder and average buried area upon folding of first 100 amino acid residue from the Translocation domain of xenocin is analyzed by Globplot and ProtScale from Expasy. Conserved amino acid residues in the putative active site of catalytic domain of xenocin have been predicted by multiple sequence alignment and surface viewed with CHIMERA software.
2. Methodology
2.1. Sequence Alignment
- The protein data banks were searched against xenocin protein, using BLASTP (http://www.ncbi.nlm.nih.gov) program. All protein sequences were obtained from NCBI sequence database (http://www.ncbi.nlm.nih.gov)
2.2. Phylogenetic Analysis
- All sequences that matched with xenocin were aligned by CLUSTALW (Mac vector 7.0), and a tree was constructed using neighbour-joining method, with the best tree mode in the Mac vector version 7.0 (Oxford Molecular, England) program.
2.3. Molecular Homology Modeling
- Three dimensional structure of xenocin was produced by using ESyPred3D (http://fundp.ac.be/urbm/bioinfo/esypred/) automated homology modeling server. Xenocin shares ~30% sequence identity with E3 colicin and as such the three dimensional structure of the latter determined by X-ray crystallography[10] was used as a specified template for homology modeling of the former in automated homology modeling. Predicted structure was verified by VERFIFY-3D (http://nihserver.mbi.ucla.edu/Verify_3D) and further analysed by Ramachandran plot 2.0 software(http://dicsoft1.physics.iisc.ernet.in/rp/select.html)
2.4. Prediction of Protein Disorder and of the Average Area Buried Upon Folding
- Protein disorder was determine by Globplot (http:// globplot.embl.de) and Prediction of the average area buried upon folding (AABUF) was calculated with the ExPASy tool ProtScale (http://us.expasy.org/tools/protscale.html)
2.5. Active site Analysis
- After the final model was built, the conserved amino acid residues in putative active site of catalytic domain of xenocin was explored multiple alignment and surface viewed with CHIMERA software (http://www.cgl.ucsf.edu/chimera)
3. Results and Discussion
3.1. Phylogenetic Analysis
- Phylogenetic analysis of the xenocin was done by preparing phylogenetic tree, using neighbor-joining method, with the best tree mode in the Mac vector version 7.0 (Oxford Molecular, England) program as shown in Figure 1. It showed that xenocin from X. nematophila formed a separate branch from other bacteria in the very beginning and was located at opposite position with reference to toxin from Klebsiella oxytoca. Toxin from Klebsiella pneumonia and E. coli cloacin DF13 form a distinct cluster where as colicin like E6 and E3 from E. coli formed their own separate cluster. Protein-protein blast showed that xenocin is just 31% similar to cloacin of K. pneumonia which is located at distal position from xenocin in phylogenetic tree however, whereas it is only 30% similar to colicin E3 which is located proximal to it phylogenetically.
3.2. Molecular Homology Modeling and Prediction of Average Area Buried
- Protein-protein blast of xenocin protein showed 30% similarity with E3 and E6 colicins. Moreover, crystal structure of E3 colicin has been resolved [10]. Therefore, three dimensional structure of xenocin was predicted by EsyPreDSD using E3 as template. Predicted structure by homology modeling consisted of a stem corresponding to receptor domain R and two globular heads of translocation domain T and catalytic domain C respectively as shown in Figure 2 (a). Analysis usingVerify3D programme showed more than 94% of the positive score value and 80% higher than 0.2. Ramachandran plot calculation showed 96.44% of the residues in the favoured and allowed regions Figure 2 (b). This analysis indicates that the model has a good quality. There are three distinct steps in the killing of the sensitive cells by colicin, each of which is carried by separate domain. The first step is receptor binding to the surface of the target cell which is mediated by the receptor domain R, which lies in the central part of the primary sequence. The N’ terminal translocation domain mediates the second step, translocation from the outer membrane receptor to the colicin’s targets in the cell. Finally, the killing activity resides in the carboxy- terminal C domain. Since, xenocin having three domain organizations; Translocation (T), Receptor (R) and Catalytic domain (C) each could have their own distinct functions.
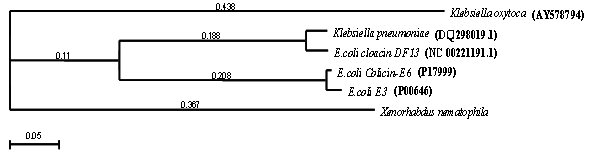 | Figure 1. Phylogenetic analysis of Xenocin with similar bacteriocins. other bacterial sources |
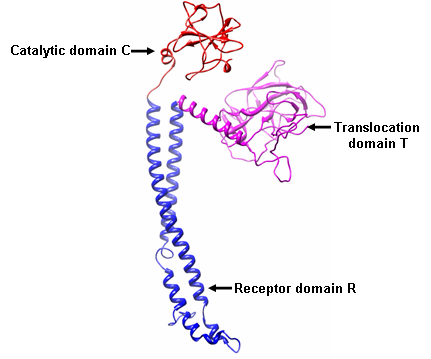 | Figure 2 (a). Three dimensional structure of Xenocin from X. nematophila predicted by automated homology modeling |
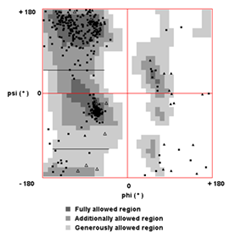 | Figure 2 (b). Validation of three dimensional structure of Xenocin from X. nematophila using Ramachandran plot |
3.2.1. Receptor Domain
- In colicin E3, receptor domain plays a major role in the first step, that is the interaction with target cell. BtuB is outer membrane protein targeted by E3 colicin. Binding of E3 with BtuB provide a scaffold for protein complex to sit on the surface of the target cells. Moreover, this interaction provides signal required for the conformational change in E3 colicin for the removal of immunity from the E3-immunity protein complex. N’ terminal of BtuB protein form a floor of a pocket that is about 25 Å deep and 25 Å in diameter. This floor is entirely made up of hydrophobic residues. The exposed hydrophobic residues of the R domain may anchor it to the floor of BtuB binding pocket[10]. In xenocin receptor domain R consists of antiparallel helical hairpin that extends from 328 to 476 amino acids as shown in Figure 3 (A) with first helical arm of this coiled coil formed by residues 328 to 407. A hairpin structure is formed by residues 408 to 413. Second helical arm extends from residues 414 to 476. In xenocin receptor domain, the upper helical region is more hydrophilic where as the lower region including the hairpin structure is mainly composed of hydrophobic amino acids as shown in Figure 3 (B and C). Most of the hydrophobic residues in these regions are exposed to the surface which may play critical and important role in interacting with receptor on the target cell.
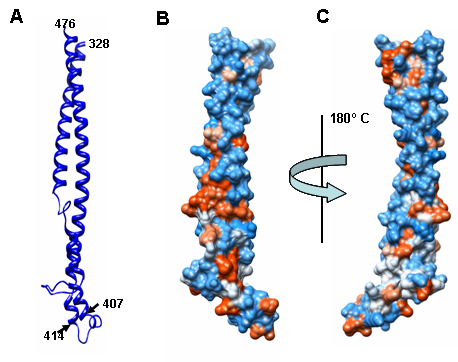 | Figure 3. Three dimensional structure of Receptor domain (R) predicted by automated homology modeling (A) Ribbon structure of receptor domain of xenocin formed by 328-476 amino acid residues. (B) & (C) Surface view of receptor domain showing the hydrophobic residues in lower position (brown color) and hydrophilic residues in the upper position (blue color) |
3.2.2. Translocation Domain
- Translocation domain in colicin is usually highly disordered in nature and this attribute helps them to interact with other outer membrane proteins. Bacteriocins are broadly classified into two groups, groups A and B, based on cross-resistance. Group A comprises of bacteriocins that are translocated by the Tol system, such as colicins A, E1 to E9, K, L, N, S4, U, and Y, while group B comprises of bacteriocins that use the TonB system, such as colicins B, D, H, Ia, Ib, M, 5, and 10[3]. N’ terminal of Tol dependent colicin contain sequence recognition motif DGSGW for the binding with TolB, a periplasmic protein involved in translocation from outer membrane to cytoplasmic membrane. Xenocin primary sequence from 1 to 327, forms N’ terminal translocation domain T. However, only 52-327 amino acid residues forms a jelly roll surface composed mainly of β sheets and α helical structures as shown in Figure 4.
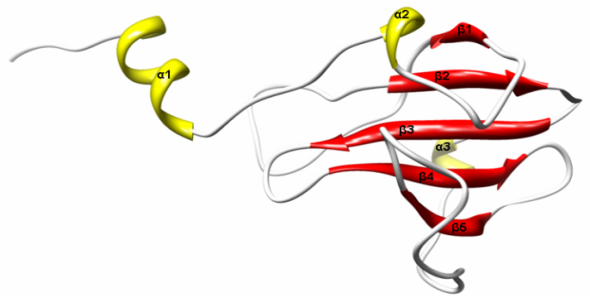 | Figure 4. Ribbon structure of Translocation domain (T) of Xenocin predicted by automated homology modeling |
3.2.3. Catalytic Domain and Active Site Prediction
- Inner membrane peptidases are important for the colicin cleavage which is necessary for killing the target cells[12]. Generally, only the catalytic domain which is resistant to proteases of inner membrane translocate to cytoplasm due to its amphiphilc nature[13]. Once they enter the cytoplasm they show detrimental effects on the target cells by targeting either DNA or RNA which ultimately inhibit the cell growth.
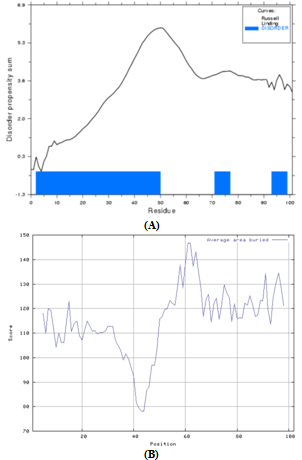 | Figure 5. (A) Disorder propensity graph for first 100 amino acid residues of xenocin predicted by Globplot 2. (B) Average area buried upon folding for first 100 first 100 amino acid residues of xenocin predicted by Protscale from Expasy |
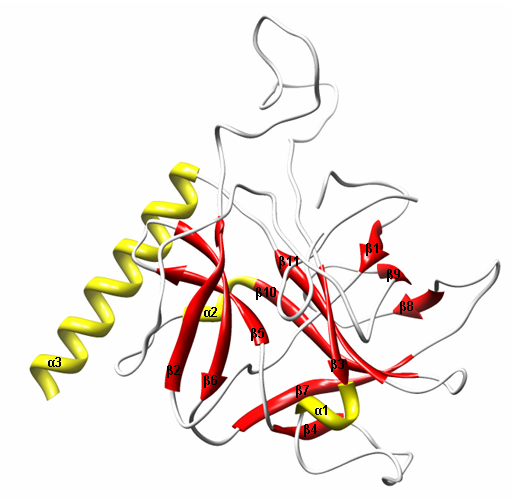 | Figure 6. Ribbon structure of catalytic domain of Xenocin predicted by automated homology modeling |
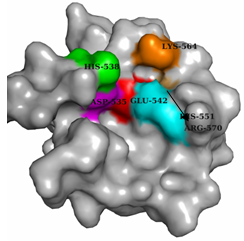 | Figure 7. Surface view of catalytic domain domain (C) of Xenocin predicted by automated homology modeling |
4. Conclusions
- The antibiotic proteins are named after the bacterial species that produce them; e.g., the “colicins” are derived from Escherichia coli[16], the “pyocins” are derived from Pseudomonas aeruginosa[17], “klebocin” is derived from Klebsiella pneumonia[18] and “xenocin” is derived from X. nematophila[9]. Most bacteriocins have a common molecular organization consisting of three functional domains, the receptor binding, membrane translocation, and cytotoxic domains. In gram-negative bacteria, the lethal action of bacteriocins involves the following essential steps. After release from the host cell, the bacteriocin binds to the specific surface receptors on the target cells, such as vitamin B12 receptor BtuB or iron siderophore receptor FepA 19 through their Receptor domain[4]. In xenocin 328-476 forms a putative receptor which may bind to the protein present on the surface of target cells. This binding is followed by import into the cells with the help of either Ton proteins (ExbB, ExbD, and TonB) or Tol proteins (TolA, -B, -Q, and -R)[2,20] for interaction with the target molecule. In our case 55-59 residues of xenocin form recognition motif specific for TonB protein, this may direct its entry into the cytoplasm by Ton pathway. Bacteriocins use variety of strategies to induce cell death through their cytotoxic domains. The pore-forming bacteriocins create voltage-gated channels, causing depolarization of the cytoplasmic membrane[21,22], while nucleases cleave tRNA or rRNA at specific sites to inhibit protein synthesis[23,10] or degrade nucleic acids nonspecifically in the target cells[24,25] In our earlier study with xenocin we have shown invitro RNase activity where as no DNAase activity had been observed. Therefore, we presume that after processing in the periplasm, only catalytic domain enters the cytoplasm of target cells and perform its detrimental effect. Conserved amino acid residues such as D535, H538, E542, R570, H551 and K564 of the catalytic domain which may involve in such activity were determined by multiple sequence alignment. Moreover, their presence on the surface to interact with negatively charged substrate is also deciphered by the surface view of all these amino acid residues. Validation of predicted amino acid residues in the putative active site of catalytic domain of xenocin, is going on with site directed mutagenesis experiments in our laboratory.