-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Bioinformatics Research
2012; 2(2): 1-10
doi: 10.5923/j.bioinformatics.20120202.01
Predicting MHC Class II Epitopes Using Separated Constructive Clustering Ensemble
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLHossam Fathy ElSemellawy 1, Amr Badr 2, Mostafa Abdel Aziem 3
1Information System Department, Arab Academy for Science, Technology and Maritime Transport, Cairo, Egypt
2Computer Science Department, Faculty of Computers and Information (Cairo University), Giza, Egypt
3Computer Science Department, Arab Academy for Science, Technology and Maritime Transport, Cairo, Egypt
Correspondence to: Hossam Fathy ElSemellawy , Information System Department, Arab Academy for Science, Technology and Maritime Transport, Cairo, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Successful Prediction for MHC Class II epitopes is an essential step in designing Genetic Vaccines[1]. MHC Class II epitopes are short peptides with length between 9 and 25 amino acids which are bound by MHC. These epitopes are recognized by T-Cell Receptors and leads to activation of cellular and humoral immune system and, ultimately, to effective destruction of pathogenic organism. Successful prediction of MHC class II epitopes is more difficult than MHC class I epitopes due to open binding groove at both ends in class II molecules, this structure leads to variable length for MHC II epitopes and complicating the task for detecting the core binding 9-mer. Large efforts have been exerted in developing algorithms to predict which peptides will bind to a given MHC class II molecules. In this paper we presented a novel classification algorithm for predicting MHC Class II epitopes using Multiple Instance Learning technique. Separated Constructive Clustering Ensemble (SCCE) is our new version for Constructive Clustering Ensemble (CCE)[27]. This method was used for converting multiple instance learning problem into normal Single Instance Problem. Most of the processing in this method lies mainly in vector preparation step before using classifier; Support Vector Machine (SVM) has been used as a method with proven performance in a wide range of practical applications[38]. SCCE integrated many algorithms like Genetic Algorithm, K medoid clustering, Ensemble learning and Support vector machine in an orchestration to predict the MHC II epitopes. SCCE was tested over three benchmark data sets and proved to be very competitive with the state of art regression methods. SCCE achieved these results using only binder and non binder flags; without need for regression data.An implementation of MHCII-SCCE as an online web server for predicting MHC-II Epitopes is freely accessible at .
Keywords: Major Histocompatibility Complex (MHC), Multiple Instance Learning (MIL), Genetic Algorithm (GA), Support Vector Machine (SVM)
Article Outline
1. Introduction
- Epitopes or antigenic peptides are set of amino acids from the pathogenic organism DNA which bound with MHC to be presented by Antigen Presenting Cells (APCs)[2] for inspection by T cells. Humans’ MHC is often called Human Leukocyte Antigen (HLA). The binding and presentation operations are considered the central recognition process occurring in the immune system as without them the immune system would be almost ineffective. These operations lead to activation of the T cell, which then signals to the wider immune system that a pathogen has been encountered.The proteins of the MHC are grouped into two classes[3]. Class I molecules present endogenous peptides, Class II molecules generally present exogenous peptides. MHC class I ligands are of 8 to 11 amino acids long while MHC class II ligands are of 9 to 25 amino acids. Class I molecules have a binding cleft which is closed at both ends while MHC class II molecules have groove which is opened at both ends this allows much larger peptides to bind and also allows the bound peptide to have significant “dangling ends”, thus the prediction of which peptides will bind a specific MHC class II complex constitutes an important step in identifying potential T cell epitopes. These epitopes are suitable as vaccine candidate. Figure 1 displays MHC II tertiary structure bounded with MHC II epitope.The currently available methods for identifying MHC II binding peptides are split into main categories:(1) Qualitative methods: These methods try to identify binder and non binder peptides despite its binding affinity, like methods use a position weight matrix to model ungapped multiple sequence alignment of MHC binding peptides[4-7], other methods use Artificial Neural Networks (ANN)[10,11] and support vector machines (SVM) like[12,13].(2) Quantitative methods: these methods try to predict binding affinity for peptide like PLS-ISC[15], MHCPred[16], SVRMHC[17], ARB[18], NetMHCII[19], MHCMIR[20] and NN-align[14].
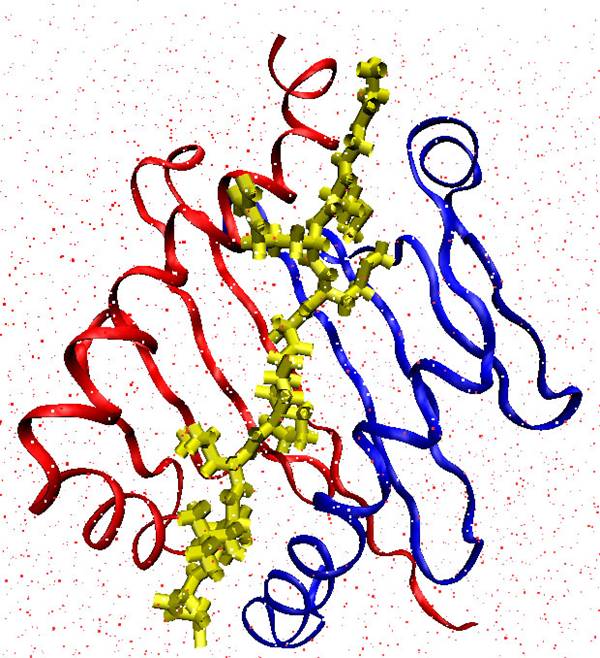 | Figure 1. Example of tertiary structure of peptide binding to MHC class II. It can be seen that the binding groove is open in the ends in contrast to MHC class I |
2. Data
- The first one is currently the largest data set which published by Wang et al.[39] for human , DP and DQ molecules binding affinities. The data set comprises 26 HLA-DR, DP and DQ alleles. Alleles included in this data set were selected for their high frequency in the human population so it reached to 99% from the human coverage.Peptides with measured IC50 < 1000 nM are considered binders and others are non binders. Wang et al. also partitioned the data into five folds used for training and testing at the following URL http://tools.immuneepitope.org/analyze/ html/download_MHC_II.html. Wang et al.[39] published comparison between set of methods by cross validation using this five folds data set. In this paper we compared the results published in Wang et al.[39] with results collected from SCCE using the same folds.The second one is IEDB HLA-DR restricted peptide-binding data set which published by Nielsen et al.[28]. As SCCE depends on qualitative input so we converted this data set from quantitative to qualitative, The peptide binding affinity IC50 has been converted to either 1 or 0 by converting all peptides with binding affinity greater than 0.426 to binder (1) and all peptides less than or equal to 0.426 to non binder. The data set comprises 13 HLA-DR alleles each characterized by at least 420 and up to 5166 peptide binding data points, only one allele data has been removed as we retrieved only allele data which have more than 100 instances in both binder and non binder class. NN-align[14] published the five folds used for training and testing on . NN-align[14] also published results on this data set for set of methods using five folds cross validation. We compared SCCE using five folded cross validation with the methods presented in NN-align[14].The third data set is El-Manzalawy benchmark data set[29]. El-Manzalawy et al. has introduced four degrees of similarity reduction for the data sets extracted from MHCPEP[40] and MHCBN[41] besides the original UPDS data set extracted from the source. El-Manzalawy et al.[29] published the results for three methods using this data set by five folds cross validation.The first degree is MHCPEP-SRDS1 and MHCBN- SRDS1 which derived from the corresponding UPDS datasets. The new data set don’t have two peptides share a 9-mer subsequence.The second degree is MHCPEP-SRDS2 and MHCBN- SRDS2 which derived from the corresponding SRDS1 datasets. The new data set ensured that the sequence identity between any pair of peptides is less than 80%.The third degree is MHCPEP-SRDS3 and MHCBN- SRDS3 which derived from the corresponding UPDS datasets using similarity reduction introduced by Raghava[42].The forth degree is MHCPEP-WUPDS and MHCBN- WUPDS which derived from the corresponding UPDS datasets, the new data set assigned weight to a peptide, this weight is inversely proportional to the number of peptides that are similar to it.The forth database is The Wang et al.[21] benchmark data set; consists of quantitative and qualitative binding data to 14 HLA-DR alleles plus IAB data set. We used this data set for comparing between the original version for CCE and our enhanced version SCCE using 10 folds cross validation.For all data sets evaluation, training data was used as an input for genetic search to find the best parameters for the training data then built ensemble classifier using these parameters and test the classifier against unseen test data.
3. Methods
3.1. Multi-Instance Learning Using Constructive Clustering Ensemble (CCE)
- The term multi-instance learning was defined by Dietterich et al.[33] when they were investigating the problem of drug activity prediction. The activity prediction objective is to predict whether a candidate drug molecule will bind strongly to a target protein or not. Not all drug molecules can bind well to all proteins. Drug molecule shape is the most important factor in determining whether a drug molecule will bind the target protein or not. However, drug molecules are flexible, so they can adopt a wide range of shapes. A binder molecule can take many shapes but at least one of them can bind to the target protein while the non binder molecule does not adapt to any shape that can bind to the protein.Multiple instance learning formulates this problem[30] by representing each candidate molecule by a bag of instances, each instance in the bag representing a unique shape adapted by the molecule. The bag is positive if and only if at least one of the instances in the bag can bind to the protein and negative if none of the instances in the bag can bind to the protein. Not like supervised learning[27] where all training instances have known labels, in multi-instance learning the labels of the training instances are unknown; and in contrast to unsupervised learning where all training instances are without known labels, in multi-instance learning the labels of the training bags are known. In multi-instance learning each instance in the bag has its own features vectors while the label is for the whole bag not for each instance.[30, 31, 32, 33] proposed a solution to the MIL problem by adapting single supervised learning algorithms to multi-instance learning as long as their focuses are shifted from the discrimination between instances to the discrimination between bags. Constructive Clustering based Ensemble (CCE) method[27] for resolving the multi-instance learning problems takes opposite direction. CCE adapted the multi-instance representation to fit with the single supervised learning algorithms, so each bag is represented in a single features vector instead of vector for each instance and this vector takes bag label.For building one vector for the whole bag instances, CCE collected all instances from all bags despite its label and placed them all in a one list, then cluster these instances into d groups. CCE represented each bag by a vector of d features (one for each cluster), the vector values are either 1 or 0; 1 if there is an instance in the bag related to this cluster and 0 if not. Now each bag is represented by only one vector containing d-dimensional binary feature vector so we could use normal single instance supervised classifiers to distinguish the bags. CCE proposed using support vector machines for classification and K-means for clustering.As there is no method for specifying the best number of clusters, CCE[27] created many classifiers using different cluster count and combined their prediction so the method utilized the power of ensemble learning to achieve strong generalization capability[34].CCE doesn’t need to store any of the training data as only clusters centroids are stored to be used for building training and testing vectors. Calculating distance between only clusters centroids and test bags instances minimizes the number of comparisons as we compare with a limited number of points not with the whole training data like Citation-kNN and Bayesian-kNN[35].CCE results were very competitive[27] whether in the MUSK data sets[33] or Generalized MI Data Sets[36] but there are some major problems in the CCE which are the following:1. There is no methodology for selecting the best number of clusters or even upper and lower boundaries.2. In case that there are shared instances between positive and negative bags which is a common case in MIL, some of the resultant clusters will be shared between positive and negative bags. These clusters will not help in distinguishing between positive and negative bags as its features have value 1 in both cases. 3. Depending on binary features vector prevented any variation in the distance between bag instances and clusters centroids.SCCE method for predicting MHC II epitopes addressed all these issues plus adapting it to fit with MHC II epitopes prediction.Comparisons between SCCE and original version for CCE are displayed to show the effect of these enhancements.
3.2. Separated Constructive clustering Ensemble Method (SCCE)
- SCCE is an enhanced version for CCE to solve multi-instance learning problem. SCCE converted bag of Multiple Instances vectors into a single vector and uses Support vector Machines (SVM) to distinguish between binder and non binder bag.First each peptide was represented by bag of 9-mer subsequences (Figure 2), then assigned a binary label whether 1 for binder bags and 0 for non binder bags. These 9-mers in each bag instances represent a candidate binding core; if the bag has at least one of these binding cores, then it is a binder bag.
 | Figure 2. representing MHC II binder peptide into bag of 9-mers subs strings, each instance is a candidate to be core 9-mers[20] |
3.3. Genetic Algorithm for Selecting Parameters
- According to previous section we need to have a methodology for specifying the parameters. The number of parameters are duplicated many times according to classifiers used in the ensemble, selecting ensemble parameters can’t be the same for all alleles specially that they vary in the number of binders and non binders, binding cores,..SCCE depends on GA for selecting ensemble parameters. First, training data were split into training and validation data set. Genetic Algorithm runs using training data and after each generation the best chromosome was tested with validation data set to make sure that the enhancements achieved in recognizing training data didn’t lead to over fitting. Once the results are going down; generations creation terminates and returns the best chromosome. Finally, we train the classifier using the whole training data with the final parameters.Genetic Algorithm chromosome contains all ensemble parameters; gene for each parameter, so for each classifier in the ensemble there is a gene for positive clusters count, negative clusters count, minimum size for positive cluster, minimum size for negative cluster and the starting position in the peptide to extract the 9-mers bag instances. GA fitness function is 3 fold cross validation result. Genetic algorithm run for specifying the parameters for 20 ensemble classifiers, we started GA with initial population of 20 randomly generated chromosomes; each chromosome has the parameters for 20 ensemble classifiers. After completing all steps for generating new generation from the initial population like selection, cross over and mutation; the best chromosome was tested using validation data; if there is an enhancements in the results then GA Search continues in generating new generation, if the result is going down on validation data set or GA search reached to maximum number of generations, GA terminates and return best chromosome.
4. Results
4.1. The Wang et al. data set[39]
- SCCE was evaluated using Wang et al.[39] data set using five folds cross validation. The data set was split into five folds and used to compare the results between several MHC Class II epitopes prediction methods. Wang et al.[39] published the results using this data set for five methods using 5-fold cross validation.SCCE classifier was created by ensemble 15 classifiers; training data has been split into training and validation. Training data was used for selecting classifiers parameters, and validation data set has been used to make sure that the new GA generations are not going to over fitting. After completing GA iterations; all training data was used for building the ensemble classifiers and tested using testing data.From the results in Table 2; SCCE is ranked number 2 after NN-align but SCCE has an advantage over NN-align which is its ability to work without need for binding affinity information. SCCE was mainly designed to work with classification data where only binary labels (binder or not binder) are available. Another advantage in SCCE is the ability to adapt it in the future to use any high generalization capability classifier rather than SVM, Currently SVM is one of the best classification methods but we can switch to any other new classifier if it proves better generalization capability.
4.2. IEDB benchmark data set
- Quantitative IEDB benchmark data set from Nielsen et al.[28] has been converted to qualitative using a threshold of 500 nM. This means that peptides with log50k transformed binding affinity values greater than 0.426 are classified as binders and peptides with binding affinity values less than or equal 0.426 as classifier as non binders. The data has been split into five folds; SCCE was evaluated using five folds cross validation.Table 3 displayed the results for SCCE using five folds cross validation with IEDB data set[28]. The first three methods results were collected from NN-align[14] using five cross validation. NN-align[14] published the five folds used for training and testing on http://www.cbs.dtu.dk/suppl/ immunology/NetMHCII-2.0.php.SCCE is very competitive with the state of art methods. Both SMM and NN-align collected its results after training using regression data while SCCE and TEPITOPE depend only on qualitative data. SCCE achieved big difference than TEPITOPE and competitive performance with SMM and NN-align without need for quantitative training data.
4.3. El-Manzalawy benchmark data set
- El-Manzalawy et al.[29] introduced new benchmark data set by using four different similarity reduction methods to create four data sets beside the original data. El-Manzalawy et al. proved that the performance for MHC II prediction method depends mainly on inherent peptide similarity in the training data and will be affected according to the similarity reduction level.SCCE performance has been compared with MHC class II epitopes prediction methods (5-spectrum, LA, and CTD) included in the El-Manzalawy et al.[29] using five folds cross validation. This comparison enabled us measure to what extend the result will be affected by similarity reduction.
|
|
|
5. Discussion
- Predicting MHC Class II binder epitopes is an essential step in developing Genetic Vaccines[1], MHC II epitopes predictions is much more complicated than predicting MHC I binders epitopes according to the open binding groove at both ends. MHC II structure leads to variable length epitopes and complicating the task for detecting the core binding 9-mer.SCCE has been inspired from both[20] and[27]. EL-Manzalawy et al.[20] has introduced converting MHC class II binding epitopes prediction into multiple instance learning; Construct Clustering Ensemble (CCE)[27] converted Multiple Instance Learning problem where each instance in the bag has its own feature vector into normal single classifier where each bag has one feature vector representing all bag instances and assign the bag label to this new feature vector. Combining both[20] and[27] enabled us to get benefit from the knowledge outside binding core and build one vector for all bag instances. SCCE has adapted CCE to MHC Class II epitopes prediction by using K-medoid clustering algorithm and BLOSUM62 amino acid substitution matrix for distance calculation.SCCE introduced separating between instances from positive and negative bags and remove shared instances between each other; then create clusters for each group separately. By this way the new clusters represent unique 9-mer in positive instances or negative instances separately and neglect representing shared 9-mers, the resultant clusters are used to build single vector for the whole bag instances. Ensemble learning has been generated between many classifiers; each one has different clusters count; by this way we used different ways for representing features and better generalization capability.SCCE also introduced using Genetic Algorithm for specifying ensemble classifiers parameters, the parameters include positive and negative clusters count and minimum cluster size. GA iterations terminate when reach to the maximum number of iterations or validation data set results is going down.SCCE used Support Vector Machine (SVM) for classification as it is currently one of the most robust methods[38] and has a very good generalization capability.
6. Conclusions
- In this paper new classification method for predicting MHC Class II epitopes was presented, this method has been tested against three main benchmark data sets and proved to be competitive with the state of art methods, SCCE is a very flexible method that can change its input vector size automatically according to patterns in training data. GA enabled SCCE to represent all feature in the training data by selecting the best number for positive and negative clusters count plus selecting the minimum cluster size. SCCE can be developed in the future to use any classifier rather than SVM or even make ensemble between SVM and any other classifiers.
ACKNOWLEDGEMENTS
- We are very Grateful to Dr Yasser EL-Manzalawy (from ) for his valuable support during our work.
References
| [1] | C. Janeway, P. Travers et al, Immunobiology: The Immune System in Health and Disease, 6th ed. Garland Pub, 2004. |
| [2] | Castellino F, Zhong G, : Antigen presentation by MHC class II molecules: invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum Immunol 1997, 54:159-169. |
| [3] | Yewdell JW, Bennink JR: Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annual review of immunology 1999, 17:51-88. |
| [4] | M. Nielsen, C. Lundegaard, and O. Lund, Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics, vol. 8, p. 238, 2007. |
| [5] | P. Reche, J. Glutting, H. Zhang, and , Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics, vol. 56, no. 6, pp. 405–419, 2004. |
| [6] | H. Singh and G. Raghava, ProPred: prediction of HLA-DR binding sites. Bioinformatics, vol. 17, no. 12, pp. 1236–1237, 2001. |
| [7] | M. Nielsen, C. Lundegaard, P. Worning, C. Sylvester-Hvid, K. Lamberth, S. Buus, S. Brunak, and O. Lund, Improved prediction of MHC class I and II epitopes using a novel Gibbs sampling approach. Bioinformatics, vol. 20, pp. 1388–97, 2004. |
| [8] | M. Rajapakse, B. Schmidt, L. Feng, and V. Brusic, Predicting peptides binding to MHC class II molecules using multi-objective evolutionary algorithms. BMC Bioinformatics, vol. 8, no. 1, p. 459, 2007. |
| [9] | H. Mamitsuka, Predicting peptides that bind to MHC molecules using supervised learning of Hidden Markov Models. PROTEINS: Structure, Function, and Genetics, vol. 33, pp. 460–474, 1998. |
| [10] | M. Nielsen, C. Lundegaard, P. Worning, S. Lauemøller, K. Lamberth, S. Buus, S. Brunak, and O. Lund, Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Science, vol. 12, pp. 1007–1017, 2003. |
| [11] | S. Buus, S. Lauemoller, P. Worning, C. Kesmir, T. Frimurer, S. Corbet, A. Fomsgaard, J. Hilden, A. Holm, and S. Brunak, Sensitive quantitative predictions of peptide-MHC binding by a’Query by Committee’ artificial neural network approach. Tissue Antigens, vol. 62, no. 5, pp. 378–384, 2003. |
| [12] | J. Cui, L. Han, H. Lin, H. Zhang, Z. Tang, C. Zheng, Z. Cao, and Y. Chen, Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. MolImmunol, 2006. |
| [13] | J. Salomon and D. Flower, Predicting Class II MHC-Peptide binding: a kernel based approach using similarity scores, BMC Bioinformatics, vol. 7, no. 1, p. 501, 2006. |
| [14] | M. Nielsen and O. Lund, NN-align. An artificial neural networkbased alignment algorithm for MHC class II peptide binding prediction. BMC bioinformatics, vol. 10, no. 1, p. 296, 2009. |
| [15] | I. Doytchinova and D. Flower, Towards the in silico identification of class II restricted Ts-cell epitopes: a partial least squares iterative self consistent algorithm for affinity prediction. pp. 2263–2270, 2003. |
| [16] | C. Hattotuwagama, P. Guan, I. Doytchinova, C. Zygouri, and D. Flower, Quantitative online prediction of peptide binding to the major histocompatibility complex. Journal of Molecular Graphics and Modelling, vol. 22, no. 3, pp. 195–207, 2004. |
| [17] | W. Liu, X. Meng, Q. Xu, D. Flower, and T. Li, Quantitative prediction of mouse class I MHC peptide binding affinity using support vector machine regression (SVR) models. BMC Bioinformatics, vol. 7, no. 1, p. 182, 2006. |
| [18] | H. Bui, J. Sidney, B. Peters, M. Sathiamurthy, A. Sinichi, K. Purton, B. Moth´e, F. Chisari, D. Watkins, and A. Sette, Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics, vol. 57, no. 5, pp. 304–314, 2005. |
| [19] | M. Nielsen, C. Lundegaard, and O. Lund: Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics, vol. 8, p. 238, 2007. |
| [20] | Yasser EL-Manzalawy, Drena Dobbs, and Vasant Honavar: Predicting MHC-II binding affinity using multiple instance regression. IEEE/ACM TRANSACTIONS ON COMPUTATIONAL BIOLOGY AND BIOINFORMATICS. |
| [21] | P. Wang, J. Sidney, C. Dow, B. Moth´e, A. Sette, and B. Peters: A Systematic Assessment of MHC Class II Peptide Binding Predictions and Evaluation of a Consensus Approach. PLoS Computational Biology, vol. 4, no. 4, 2008. |
| [22] | C. Lawrence, S. Altschul, M. Boguski, J. Liu, A. Neuwald, and J. Wootton: Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science, vol. 262, no. 5131, pp. 208–214, 1993. |
| [23] | S. Chang, D. Ghosh, D. Kirschner, and J. Linderman: Peptide length based prediction of peptide-MHC class II binding. Bioinformatics, vol. 22, no. 22, p. 2761, 2006. |
| [24] | Y. Chen, J. Bi, and J. Wang, MILES: Multiple-instance learning via embedded instance selection. IEEE Trans Pattern Anal Mach Intell, vol. 28, no. 12, pp. 1931–1947, 2006. |
| [25] | S. Henikoff and J. Henikoff, Amino Acid Substitution Matrices from Protein Blocks. Proceedings of the National Academy of Sciences of the United States of America, vol. 89, no. 22, pp. 10 915–10 919, 1992. |
| [26] | S. Shevade, S. Keerthi, C. Bhattacharyya, and K. Murthy: Improvements to the SMO Algorithm for SVM Regression. IEEE Transactions on Neural Networks, vol. 11, no. 5, p. 1189, 2000. |
| [27] | Zhi-Hua Zhou, Min-Ling Zhang: Solving Multi-Instance Problems with Classifier Ensemble Based on Constructive Clustering. Knowledge and Information Systems, 11(2):155-170, 2007. |
| [28] | Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O: Quantitative predictions of peptide binding to any HLADR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol 2008, 4(7):e1000107. |
| [29] | El-Manzalawy Y, Dobbs D, Honavar V: On evaluating MHC-II binding peptide prediction methods. PLoS One 2008, 3(9):e3268. |
| [30] | R. H. Dietterich, T. G.; Lathrop and T. Lozano-Perez, Solving the multiple-instance problem with axis parallel rectangles. Artificial Intelligence, vol. 89(1-2), pp. 31–71, 1997. |
| [31] | Zhou Z-H, Zhang M-L (2003) Ensembles of multi-instance learners. In Lavra·c N, Gamberger D, Blockeel H, Todorovski L (eds). Lecture Notes in Artificial Intelligence 2837, Springer, , pp 492-502. |
| [32] | S. Andrews, I. Tsochantaridis, and T. Hofmann, Support vector machines for multiple-instance learning. Advances in Neural Information Processing Systems, vol. 15, 2002. |
| [33] | T. Gartner, P. Flach, A. Kowalczyk, and A. Smola, Multi-instance kernels. Proceedings of the 19th International Conference on Machine Learning, pp. 179–186, 2002. |
| [34] | Dietterich TG (2000) Ensemble methods in machine learning. In Kittler J, Roli F (eds). Lecture Notes in Computer Science 1867, Springer, , pp 1-15. |
| [35] | Wang J, Zucker J-D (2000) Solving the multiple-instance problem: A lazy learning approach. In Proceedings of the 17th International Conference on Machine Learning, , 2000, pp 1119-1125. |
| [36] | Weidmann N, Frank E, Pfahringer B (2003) A two-level learning method for generalized multi-instance problem. In Lavra·c N, Gamberger D, Blockeel H, Todorovski L (eds). Lecture Notes in Artificial Intelligence 2837, Springer, , pp 468-479. |
| [37] | Jiawei Han & Micheline Kamber (2001), Data Mining Concepts and Techniques, pp351. |
| [38] | Cristianini N, Shawe-Taylor J: An introduction to support vector machines and other kernel-based learning methods. , , Press; 2000. |
| [39] | Peng Wang, John Sidney, Yohan Kim, Alessandro Sette, Ole Lund, Morten Nielsen, Bjoern Peters: Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 2010, 11:568. |
| [40] | Brusic V, Rudy G, Harrison L, Journals O. MHCPEP a database of MHCbinding peptides: update 1997. Nucleic Acids Res 26: 368–371. |
| [41] | Bhasin M, Singh H, Raghava G (2003) MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics 19: 665–666. |
| [42] | Raghava G. MHCBench: Evaluation of MHC Binding Peptide Prediction Algorithms. Available athttp://www.imtech.res.in/raghava/mhcbench/. |