R. F. Mansour
Department of Science and Mathematics, Faculty of Education, New Valley, Assiut University, EL kharaga, Egypt
Correspondence to: R. F. Mansour , Department of Science and Mathematics, Faculty of Education, New Valley, Assiut University, EL kharaga, Egypt.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Abstract
Protein structure prediction (PSP) has important applications in different fields, such as drug design, disease prediction, and so on. In protein structure prediction, there are two important issues. The first one is the design of the structure model and the second one is the design of the optimization technology. Because of the complexity of the realistic protein structure, the structure model adopted in this paper is a simplified model, which is called off-lattice AB model. After the structure model is assumed, optimization technology is needed for searching the best conformation of a protein sequence based on the assumed structure model. However, PSP is an NP-hard problem even if the simplest model is assumed. Thus, many algorithms have been developed to solve the global optimization problem. In this paper, a hybrid algorithm, which combines genetic algorithm based on matrix coding (GAMC) and tabu search (TS) algorithm, is developed to complete this task. Experiments are performed with Fibonacci sequences and real protein sequences. Results show that the lowest energy obtained by the proposed GTAMC algorithm is lower than that obtained by previous methods. Our algorithm has better performance in global optimization and can predict 3D protein structure more effectively.
Keywords:
Energy Minimization; Protein Structure Prediction; Optimization and Off-Lattice Model
Cite this paper:
R. F. Mansour , "Applying an Evolutionary Algorithm for Protein Structure Prediction", American Journal of Bioinformatics Research, Vol. 1 No. 1, 2011, pp. 18-23. doi: 10.5923/j.bioinformatics.20110101.03.
1. Introduction
Protein structure prediction is defined as the prediction of the tertiary structure of a protein by using its primary structure information. It has become an important research topics in bioinformatics and it has important applications in medicine and other fields, such as drug design, prediction of diseases, and so on. Because of the complexity of the realistic protein structure, it is hard to determine the exact tri-dimensional structure from its sequence of amino acids[1]. Therefore, a lot of coarse structure models have been developed. The HP model is the most conventional one among them and has been widely used in protein structure prediction. Different from the complex structure models, HP model only assumes two types of amino acids-hydrophobic (H) and hydrophilic (P) and the sequence of amino acids is assumed to be embedded in a lattice, which is used to discretize the space of conformations. For simplicity, the only interaction considered in HP model is the interaction between the nonadjacent but next-neighbored hydrophobic monomers, which is used to force the formation of a compact hydrophobic core as observed in real proteins[2]. Although simplified models have the capability of catching nontrivial aspects of the folding problem, the approximations involved are not reallysuitable[3].The main reason lies in that local interactions are neglected in the simplified models. As is well known, local interactions might be important for the local structure of the chains[4] and no sequences with compact, well-defined native structures could be found if local interactions are neglected[3]. Therefore, many other models which consider local interactions have drawn a lot of attention and been proposed. The AB off-lattice model is the one that could meet the aforementioned requirement. Currently, AB off-lattice model has been widely applied to protein structure prediction and many improved models have been proposed based on the original model. In AB off-lattice model, two types of monomers are taken into consideration. The hydrophobic monomers are labelled by A while the hydrophilic ones are labelled by B. Different from HP model, the interactions considered in AB model include both sequence independent local interactions and the sequence dependent Lennard- Jones term that favors the formation of a hydrophobic core. After a structure model is adopted, an important issue in PSP is to develop an optimization technology to find the best conformation of a protein sequence based on the assumed structure model. However, protein structure prediction (PSP) is an NP-hard problem even when the simplest models are assumed[5,6].In order to tackle this issue, many heuristic approaches have been developed. In the past decades, researchers have developed many algorithms to solve the global optimization problem in protein folding structure prediction (PFSP). Genetic algorithm has been used for protein structure prediction for long time[7,10]. The reason why Gas are attractive is possibly due to their simplicity and efficiency in finding good solutions in large and complex search spaces. It is well known that the combination of GA with local search strategies is particularly effective in PFP[1]. For example, the algorithm developed in[11] which is a hybrid scheme combining GA with simulated annealing algorithm, has much higher efficiency in searching for native states with off-lattice AB model than other methods. However, this method has a limitation that the searching time is too long, which affects its wide applications. In this paper we propose a novel hybrid approach for protein structure prediction. The proposed algorithm will combine genetic algorithm and tabu search algorithm to accurately search for the ground state conformation of a given protein.
2. The Proposed Method
2.1. Off-lattice AB Model
The off-lattice AB model has been applied to protein structure prediction for decades. In off-lattice AB model, the monomers are linked by rigid unit-length bonds to form linear unoriented polymers in three-dimensional space. The energy functional for any n monomers chain is described as follows[12]: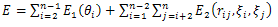 | (1) |
Where 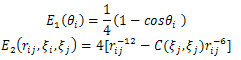 where θi (0≤θi≤π) is the angle between two successive bond vectors. rij is the distance between residues i and j with i <j. In three-dimensional space, rij depends on both bond angle θ and torsional angle β .In three-dimensional space, rij depends on both bond angle θ and torsional angle β. The constant C(ξi, ξj) is given as follows:
where θi (0≤θi≤π) is the angle between two successive bond vectors. rij is the distance between residues i and j with i <j. In three-dimensional space, rij depends on both bond angle θ and torsional angle β .In three-dimensional space, rij depends on both bond angle θ and torsional angle β. The constant C(ξi, ξj) is given as follows: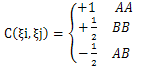 | (2) |
In off-lattice AB model, the shape of an n-mer is determined by the (n-2) bond angles θ1,…,θn-2, and the (n-3) torsional angles β1,…,βn-3. Therefore, the prediction of 3D folding structure problem of n monomers chain is equivalent to finding the optimal (n-2) bond angles and (n-3) torsional angles which minimize the energy functional E defined in equation (1).
2.2. Improved Genetic Tabu Algorithm
Genetic algorithms are adaptive heuristic search algorithms premised on the evolutionary ideas of natural selection and genetics, which select individuals by a fitness function. Individuals with higher fitness values have higher opportunity to generate the successors. Although genetic algorithms are widely used in optimization problems, they still need improvement for PSP. GA has main disadvantage which affect their performance for PSP. The disadvantage is the premature convergence and the other is the slow convergence rate, and the premature convergence is mainly caused by the small variability in mutation strategy. In order to overcome the disadvantages in GAs, we introduce tabu search (TS)[13] into the mutation operator in GAs to improve the local search capability. Tabu search is a local neighborhood search algorithm which guides the next search direction by using flexible memory functions to record and choose the optimization process. The advantage of TS is the short searching time and the disadvantage is the low global search capability. Thus, the combination of GA and TS results in a hybrid algorithm which combines both of the advantages of the GA and TS[13]. The following five strategies are used in the proposed algorithm for protein structure prediction:
2.2.1. Chromosome Encoding
Chromosome encoding is the way the individuals are represented and is very important because it affects the performance of a genetic algorithm. In the proposed algorithm, Cartesian coordinates are adopted to represent the individuals because of its simplicity. Let h be an individual. For an n-residue long chain, h can be expressed as (θ1,…,θn-2, β1,…,βn-3), which concatenates the (n-2) bond angles and the (n-3) torsional angles. Cartesian coordinates of residue i in hypothesis h(θ1 …,θn-2,β1,…,βn-3) is obtained as follows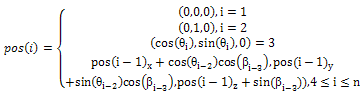 | (3) |
The coordinates of the first few residues are (0,0,0), (0,1,0), and (cos(θ1),sin(θ1),0). Latter residues’ coordinates are all calculated on the base of the previous one’s coordinate.
2.2.2. Variable Population Size
In genetic algorithm, with the difference between individuals get smaller and smaller after several rounds of evolution, premature convergence to poor solution will generally happen. Hence, the new strategy used in the proposed algorithm is to adopt variable population size. Variable population size strategy adopted by genetic algorithm can prevent premature convergence by increasing or decreasing the population size when the optimal energy is very close to the average value of the population .The proposed method starts with an initial population P0 of size μ. This initial population is coded as a matrix of size μ× n called initial population matrix PM0. At every generation t, PMt is partitioned into v × η sub- matrices 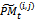 i = 1, ..., v, j = 1, ..., η , where v is the number of individuals on each partition and η is the number of genes on each partition. A crossover and mutation operators are applied on the partitioned sub-matrices and
i = 1, ..., v, j = 1, ..., η , where v is the number of individuals on each partition and η is the number of genes on each partition. A crossover and mutation operators are applied on the partitioned sub-matrices and 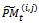 is updated. The range of each gene is divided into m sub-ranges in order to check the diversity of the gene values. The Gene Matrix GM[14] is initialized to be the n × m zero matrix in which each entry of the i-th row refers to a sub-range of the i-th gene. While the search is processing, the entries of GM are updated if new values for a gene are generated within the corresponding sub-range. After having a GM full, i.e., with no zero entry, the search is learned that an advanced exploration process has been achieved. The termination criteria in the GA is based on the GM presented in[14].
is updated. The range of each gene is divided into m sub-ranges in order to check the diversity of the gene values. The Gene Matrix GM[14] is initialized to be the n × m zero matrix in which each entry of the i-th row refers to a sub-range of the i-th gene. While the search is processing, the entries of GM are updated if new values for a gene are generated within the corresponding sub-range. After having a GM full, i.e., with no zero entry, the search is learned that an advanced exploration process has been achieved. The termination criteria in the GA is based on the GM presented in[14].
2.2.3. Crossover
Arithmetical crossover operation is used in the proposed algorithm to crossover operation between individuals from the current population into next generation. We can defined in the following procedure.Procedure Crossover(p1, p2)1- Choose randomly a number α from (0,1)2- Two offspring 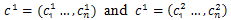 are generated from parents
are generated from parents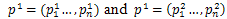 where
where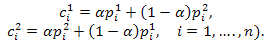 3- Return.
3- Return.
2.2.4. Tabu Search Mutation
In the proposed algorithm, the mutation operator adopted is tabu search mutation operator. Tabu search mutation operator is similar to the standard mutation operator except that TSM is a search process. With this strategy, the potential energy functional in equation (1) is used as the evaluation function to compute the offspring’s energy values, and then these offspring and their energy values are combined with the tabu list to determine the output offspring. Therefore, TSM can accept inferior solutions during the search process, and thus it has stronger hill-climbing capability than many other mutation operators[15]. TSM is composed of several steps, which can be described as follows: Firstly, disturbance mutation method is used to generate neighbor solutions of the current solutions. In this processing, two mutation operations are used. The first mutation operation is a two-point mutation operation and is used in the early stage; the second mutation operation is a single-point mutation which is adopted in the later stage to raise the convergence speed. Disturbance mutation implementation is presented as follows. Let the j th parameter selected be 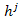 and the new parameter be
and the new parameter be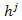 , then we have
, then we have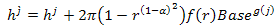 | (4) |
where r is a random number between 0 and 1. α ∈[0,1] in term. 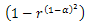 is used to assure large disturbance degree in the early search procedure (a tends to 0) to keep the diversity of the solutions, and small disturbance degree in the later search procedure (α tends to 1) to increase convergence rate and guarantee the algorithm to converge to a global optimum. f (r)and g( j) are defined as follows:
is used to assure large disturbance degree in the early search procedure (a tends to 0) to keep the diversity of the solutions, and small disturbance degree in the later search procedure (α tends to 1) to increase convergence rate and guarantee the algorithm to converge to a global optimum. f (r)and g( j) are defined as follows: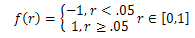 | (5) |
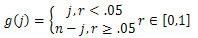 | (6) |
Baseg()j is used to ensure the diversity of the neighbor solutions, which is similar to[16]. j donates the location of the j th parameter in individual h, n is the parameter length of h. Base∈[0.9,0.99] is the scale factor of parameter hj. Secondly, the individuals in the neighbor solutions are sorted by the energy values in ascending order and the lower energy individuals will be used to generate the candidate set. Finally, each solution in the candidate set will be determined to be the output of the TSM or not. This processing is based on two tabu lists as in[17].The first tabu list is composed of a set of solution vectors and the second one is composed of a set of energy values of the corresponding solutions. The use of two tabu lists can let the algorithm avoid being trapped in local optima. In order to determine whether a candidate solution is a tabu, we use the following criteria: let the energy value of the candidate solution h(θ1,…,θn-2, β1,…,βn-3). be E(h) computed by (1)). If there is a solution y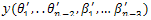 in tabu list TS which satisfies
in tabu list TS which satisfies 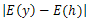 ≤ ψ and ||y-h|| ≤ η, then the candidate solution h is thought as a tabu. In TSM, it is possible that total tabu[17] happens. When total tabu happens, all the solutions in the candidate set are forbidden and the next current solution cannot be selected from the candidate set. In order to handle total tabu, when total tabu happens, we select the global best solution to generate mutation and the output solution will be set as current solution.
≤ ψ and ||y-h|| ≤ η, then the candidate solution h is thought as a tabu. In TSM, it is possible that total tabu[17] happens. When total tabu happens, all the solutions in the candidate set are forbidden and the next current solution cannot be selected from the candidate set. In order to handle total tabu, when total tabu happens, we select the global best solution to generate mutation and the output solution will be set as current solution.
2.2.5. Tabu Search Recombination (TSR)
Another strategy used in the proposed algorithm is tabu search recombination. With this strategy, TSR records the fitness values of individuals in a tabu list and the fitness values of the offspring individual after crossover operation will be compared with some desired level and will be determined to be accepted by next generation or the tabu list. In this paper, the average fitness value of the population is considered as the desired level and the crossover strategy is random linear combination. Let h(θ1,…,θn) be one individual and 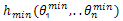 be the other individual of the crossover couple,
be the other individual of the crossover couple, 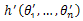 be the offspring individual after crossover operation, random linear combination is described by[18]
be the offspring individual after crossover operation, random linear combination is described by[18]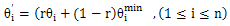 | (7) |
Where 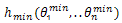 is the individual with minimum energy so far. In order to avoid close match, before the crossover operation, TSR uses the following way to select the individuals as the crossover individual couple into cross pool: it will first sort the individuals by their energy values in ascending order, and then the individuals with the furthest distance are selected for crossover operation. After the crossover offspring
is the individual with minimum energy so far. In order to avoid close match, before the crossover operation, TSR uses the following way to select the individuals as the crossover individual couple into cross pool: it will first sort the individuals by their energy values in ascending order, and then the individuals with the furthest distance are selected for crossover operation. After the crossover offspring 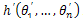 is obtained, its fitness value is compared with the desired level. The comparison is as follows: if the fitness value of the offspring is better than the desired value, the offspring will be set free, and accepted by next generation. If the fitness value of the offspring is worse than the desired value and is not in tabu list, the offspring will also be accepted. However, if the offspring is in the tabu list, TSR will select one parent with better fitness value to go into the next generation. In TSR, the use of tabu list keeps the diversity among individuals and avoids premature convergence.
is obtained, its fitness value is compared with the desired level. The comparison is as follows: if the fitness value of the offspring is better than the desired value, the offspring will be set free, and accepted by next generation. If the fitness value of the offspring is worse than the desired value and is not in tabu list, the offspring will also be accepted. However, if the offspring is in the tabu list, TSR will select one parent with better fitness value to go into the next generation. In TSR, the use of tabu list keeps the diversity among individuals and avoids premature convergence.
3. Genetic and Tabu Algorithm Based on Matrix Coding (GTAMC)
Genetic algorithm and tabu search algorithm have their own advantages and disadvantages, thus the development of a scheme which keeps the advantages while overcomes the disadvantages of each algorithm can provide efficient search for protein structure prediction. GTAMC, a hybrid algorithm, satisfies this requirement. For example, GTAMC makes use of the advantages of multiple search points in GA and can overcome the poor hill-climbing capability by using the flexible memory functions of TS. The search algorithm starts with the initialization of parameters by some appropriate values. Then, the population P with individuals h(θ1,…,θn-2, β1,…,βn-3) is generated randomly, and equation (1) is used to obtain the energy values. After that, the individuals are sorted by the energy values from minimum to maximum and at the same time, the minimal solution and the minimal energy are saved as hmin and Emin respectively. During the search process, population P is handled by TSR and TSM by turns. When TSR handles the population, it will select r*n parents from the latter 90% locations to perform crossover operation with hmin, and the preceding 10% locations are recognized as duplicated individuals. The offspring will be considered whether are accepted based on the current tabu list. When TSM handles the population, m*n mutation parents will be selected probabilistically, and each parent uses TSM operation to generate offspring. Whenever the population P is updated, individuals will be rearranged to be from minimal to maximal by the energy values. Finally, the hypothesis hmin and minimum energy Emin will be used as the optimal values at the end of algorithm.The formal detailed description of GTAMC is given in the following algorithm.1- Initialization Set values of m, μ, v and η . Set the crossover and mutation probabilities pc 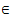 (0,1) and pm
(0,1) and pm 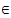 (0,1), respectively. Set the generation counter t := 0. Initialize GM as n × m zero matrix, and generate an initial population P0 of size μ and code it to a matrix
(0,1), respectively. Set the generation counter t := 0. Initialize GM as n × m zero matrix, and generate an initial population P0 of size μ and code it to a matrix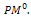 2- Parent selection. Evaluate the fitness function of all individuals coded in
2- Parent selection. Evaluate the fitness function of all individuals coded in 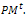 Select an intermediate population
Select an intermediate population 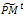 from the current one
from the current one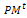 . 3- Partitioning and genetic operations. Partition
. 3- Partitioning and genetic operations. Partition 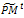 into v × η sub-matrices. Apply the following for all sub-matrices
into v × η sub-matrices. Apply the following for all sub-matrices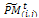 , i = 1, ..., η, j = 1, ..., v.i. Crossover. Associate a random number from (0, 1) with each row in
, i = 1, ..., η, j = 1, ..., v.i. Crossover. Associate a random number from (0, 1) with each row in 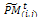 and add this individual to the parent pool if the associated number is less than pc. Apply Procedure crossover to all selected pairs of parents and update
and add this individual to the parent pool if the associated number is less than pc. Apply Procedure crossover to all selected pairs of parents and update 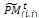 ii. Mutation. Associate a random number from (0, 1) with each gene in each gene i
ii. Mutation. Associate a random number from (0, 1) with each gene in each gene i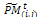 . Mutate the gene which their associated number less than pm by generating a new random value for the selected gene within its domain.4- Stopping condition. If GM is full, then go to Step 7. Otherwise, go to Step 5.5- Survivor selection. Evaluate the fitness function of all corresponding children in
. Mutate the gene which their associated number less than pm by generating a new random value for the selected gene within its domain.4- Stopping condition. If GM is full, then go to Step 7. Otherwise, go to Step 5.5- Survivor selection. Evaluate the fitness function of all corresponding children in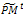 , and choose the μ best individuals from the parent and children populations to form the next generation
, and choose the μ best individuals from the parent and children populations to form the next generation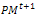 .6- Mutagenesis.7- For (loopCounter:=0, loopCounter ++<GA Maxloop) applying {TSRTSM}and go to Step 2.8- Intensification. Apply a local search method starting from each solution from the Nelite elite ones obtained in the previous search stage.
.6- Mutagenesis.7- For (loopCounter:=0, loopCounter ++<GA Maxloop) applying {TSRTSM}and go to Step 2.8- Intensification. Apply a local search method starting from each solution from the Nelite elite ones obtained in the previous search stage.
4. Results and Discussion
4.1. Results for Fibonacci Sequences
In this section, we describe our experiments by using Fibonacci sequences to test the efficiency of the proposed GTAMC. A Fibonacci sequence is defined recursively by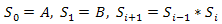 | (8) |
Where * is the concatenation operator. Some examples of Fibonacci sequences are S2 = AB, S3 = BAB, S4 =ABBAB, etc. For comparison, we used the same Fibonacci sequences as those used in[16,19-21]. GTAMC was implemented by Matlab program in Windows XP. The parameters in the algorithm were obtained by experiments and they were set as follows: self-adjustable population scale was set to be in the range of 100~500, the crossover rate was set to be 0.88, the mutation rate was set to be in the range of 0.012~0.025, self-adjustable tabu list length was set to be in the range of 7~14, neighborhood set length was set to be in the range of 30~50, candidate set length was set to be in the range of 5~6. The minimal energy values (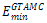 ) obtained by GTAMC on the three-dimensional off-lattice AB model are listed in Table 1. For comparison, we also list the minimal energy values obtained by the Simulated Annealing (SA)[21].
) obtained by GTAMC on the three-dimensional off-lattice AB model are listed in Table 1. For comparison, we also list the minimal energy values obtained by the Simulated Annealing (SA)[21].| Table 1. Lowest energies for Fibonacci sequences obtained by the previous algorithms and the proposed GTAMC algorithm. |
| | N | Sequences |
|
|
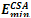
|
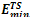
|
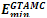
| | 13 | ABBABBAB ABBAB | -4.9746 | -4.967 | -4.97461 | -6.5687 | -6.95739 | | 21 | BABABBABAB BABBABABBAB | -12.0617 | -12.316 | -12.3266 | -13.4151 | -14.2984 | | 34 | ABBABBABABB ABBABABBABA BBABBABABBAB | -23.0441 | -25.476 | -25.5113 | -27.9903 | -28.6376 | | 55 | BABABBABA BBABBABAB BABABBABBAB ABBABBABA BBABABBABBA BABBAB | -38.1977 | -42.428 | -42.3418 | -41.5098 | -42.5936 |
|
|
The energy landscape paving minimize (ELP)[19], the conformational space annealing (CSA)[19], and the tabu search algorithm (TS) respectively. From table 1, we can find that the lowest energy value 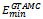 obtained by the proposed GTAMC is smaller than those obtained by SA, ELP and CSA for all the four Fibonacci sequences, and smaller than that obtained by TS for the sequences with lengths 13, 21, 55. Although the lowest energy value obtained by GTAMC is not as low as that obtained by TS for the sequence with length 34, it is smaller than that obtained by TS for the sequences with lengths 55, which shows that GTAMC has better performance for long sequence. The lowest-energy ground configurations of Fibonacci sequences listed in Table 1 are presented in Figure 3. Shows that all the conformations form single compact hydrophobic cores surrounded by hydrophilic residues, which is observed in real proteins. The results verify that it is reasonable to use AB model with Fibonacci sequences in three dimensions to mimic the real protein. In Figure 3 shows the initial random conformation, which generated by GTAMC at n = 13, 21, 34 and 55. The red balls represent hydrophobic A monomers, and the gray balls represent hydrophilic B monomers. Figure 3 shows that the results are unstable.
obtained by the proposed GTAMC is smaller than those obtained by SA, ELP and CSA for all the four Fibonacci sequences, and smaller than that obtained by TS for the sequences with lengths 13, 21, 55. Although the lowest energy value obtained by GTAMC is not as low as that obtained by TS for the sequence with length 34, it is smaller than that obtained by TS for the sequences with lengths 55, which shows that GTAMC has better performance for long sequence. The lowest-energy ground configurations of Fibonacci sequences listed in Table 1 are presented in Figure 3. Shows that all the conformations form single compact hydrophobic cores surrounded by hydrophilic residues, which is observed in real proteins. The results verify that it is reasonable to use AB model with Fibonacci sequences in three dimensions to mimic the real protein. In Figure 3 shows the initial random conformation, which generated by GTAMC at n = 13, 21, 34 and 55. The red balls represent hydrophobic A monomers, and the gray balls represent hydrophilic B monomers. Figure 3 shows that the results are unstable.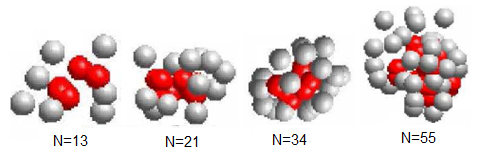 | Figure 3. The initial conformations obtained by GTAMC algorithm. |
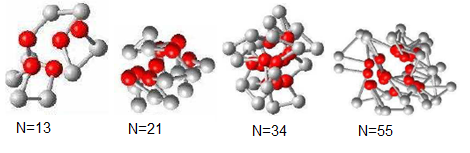 | Figure 4. The lowest energy conformations for the four Fibonacci sequences obtained by GTAMC algorithm. |
However Figure 4 shows that the lowest energy conformations in the 3D off-lattice model obtained by GTAMC algorithm, corresponding to the energies shown in Table 1. This indicates that the AB model in three dimensions with Fibonacci sequences displays the important feature when it is used to simulate the real proteins.
4.2. Results for Real Protein Sequences
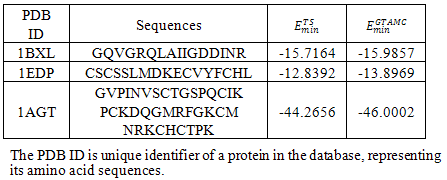
In this section, we describe the experimental results using real protein sequences. The real protein sequences used in our experiments were downloaded from the website: http://pdbbeta.rcsb.org/pdb/Welcome.do. For comparison, we used the same three protein sequences as those used in[22]. The PDB ID of the three protein sequences are 1BXL, 1EDP and 1AGT, respectively. In the experiments, the same K-D method used in[22,23] were adopted to distinguish the hydrophobic monomers from the hydrophilic ones, where I, V, L, P, C, M, A, G are considered to be hydrophobic while D, E, F, H, K, N,Q, R, S, T, W, Y are hydrophilic. Because there are few papers dealing with the real protein structure prediction issue using off-lattice AB model, we only compared our experimental results with the results in[22].Table 2. Minimum energies for three real proteins obtained by TS and GTAMC algorithm using off-lattice AB model in three dimensions.
 |
| |
|
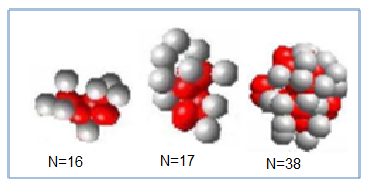 | Figure 5. shows the initial conformation of the real proteins, which generated by GTAMC. |
The experimental results for the real proteins are presented in Table 2 and the corresponding lowest protein landscapes obtained by our GTAMC are shown in Figure 5 and 6. Table 2 shows that the minimal energy values obtained by the proposed GTAMC are lower than those obtained by TS in[22], especially for long sequences. The results demonstrate that GTAMC is much more efficient than TS in protein folding structure prediction using AB off-lattice model. Figure 5 shows the initial conformation of the real proteins, which generated by GTAMC at n = 16, 17 and 38.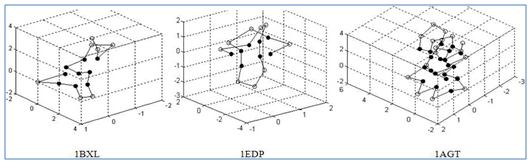 | Figure 6. The lowest energy conformations for the three real protein sequences obtained by GATS algorithm Solid dots indicate hydrophobic monomers I, V, L, P, C, M, A, G, and open dots indicate hydrophilic monomers D, E, F, H, K, N, Q, R, S, T, W, Y. |
From Figure 6, we find that all the configurations have also formed a hydrophobic core, surrounded by hydrophilic residues. However, the hydrophobic core of 1AGT, which is the longest among the three real proteins, seems not to be compact enough. This may indicate that the performance of the coarse simplified AB off-lattice model is not effective enough for the prediction of the structure for long protein sequences
5. Conclusions
A hybrid algorithm that combines genetic algorithm and tabu search algorithm is developed for 3-D protein structure prediction using off-lattice AB model. The proposed algorithm can deal with multi-parameter problems. In the proposed algorithm, different strategies are adopted to make the proposed algorithm have different advantages. For examples, the variable population size strategy can keep the diversity of the population, and TSM strategy makes it possible to accept poor solution as the current solution and thus makes the algorithm have better hill-climbing capability and stronger local searching capability than many other mutation operators. In addition, TSR strategy can limit the frequency that the offsprings with the same fitness appear, and thus can also keep the diversity of the population and avoid premature convergence of the algorithm. Compared with the previous algorithms, GTAMC has stronger capability of global searching. In the future work, we will improve the algorithm and make it more effective for long protein sequence prediction using multi-core computing platforms[24].
References
| [1] | Lopes HS: Evolutionary Algorithms for the Protein Folding Problem: A Review and Current Trends. Studies in Computational Intelligence Springer Berlin 2008, 151:297-315 |
| [2] | Hart WE, Newman A: Protein structure prediction with lattice models. Handbook of Molecular Biology CRC Press Aluru S. Chapman & Hall/CRC Computer and Information Science Series 2006, 1-24 |
| [3] | Irbäck A, Sandelin E: Local Interactions and Protein Folding: Model Study on the Square and Triangular Lattices. J. Chem. Phys. 1998, 108(5):2245-2250 |
| [4] | Irbäck A, Peterson C, Potthast F, Sommelius O: Local interactions and protein folding: A three-dimensional off-lattice approach. J. Chem. Phys. 1997, 107:273-282 |
| [5] | Hart WE, Istrail S: Robust proofs of NP-hardness for protein folding general lattices and energy potentials. Journal of Computational Biology 1997, 4(1):1-22 |
| [6] | Ngo JT, Marks J, Karplus M: Computational complexity, protein structure prediction, and the Levinthal paradox. The Protein folding problem and tertiary structure prediction Mertz M, Grand ML. S Birkhauser 1994, 433-506 |
| [7] | Hoque MT, Chetty M, Dooley LS: A New Guided Genetic Algorithm for 2D Hydrophobic-Hydrophilic Model to Predict Protein Folding. IEEE Congress on Evolutionary Computation 2005 |
| [8] | Corne DW, Fogel GB: An Introduction to Bioinformatics for Computer Scientists. Evolutionary Computation in Bioinformatics Elsevier IndiaFogel GB, Corne DW 2004, 3-18 |
| [9] | Takahashi O, Kita H, Kobayashi S: Protein Folding by a Hierarchical Genetic Algorithm. 4th Int. Symp. AROB. 1999, 19-22 |
| [10] | König R, Dandekar T: Refined Genetic Algorithm Simulation to Model Proteins. Journal of Molecular Modeling Springer Berlin 1999, 5:317-324 |
| [11] | Zhang X, Lin X, Wan C, Li T: Genetic-Annealing Algorithm for 3D Off lattice Protein Folding Model. PAKDD workshops 2007, 4819:186-193 |
| [12] | Zhang et al3D Protein structure prediction with genetic tabu search algorithm, BMC Systems Biology, 4(Suppl 1):S6, 2010. |
| [13] | Glover F, Kelly JP, Laguna M: Genetic algorithms and tabu search: Hybrids for optimization. Computers and Operations Research 1995, 22(1):111-134 |
| [14] | Hedar, B. T. Ong and M. Fukushima, “Genetic algorithms with automatic accelerated termination,” Technical Report 2007-002, Department of Applied Mathematics and Physics, Kyoto University, (January 2007) |
| [15] | Zhu J: Non-classical mathematics for Intelligent Systems. Huazhong University of Science and Technology Press 2001, 285-288 |
| [16] | Li D, Wang L, Wang M: Genetic algorithms and tabu search: a hybrid strategy. Journal of Systems engineering 1998, 13(3):28-34 |
| [17] | Zhang X, Cheng W: Protein 3D structure prediction by improved tabu search in off-lattice AB model. ICBBE 2008, 184-187 |
| [18] | Michalewicz Z: Genetic algorithms + data structures = evolution programs. Springer-Verlag, 3rd 1996 |
| [19] | Bachmann M, Arkin H, Janke W: Multicanonical study of coarse-grained off-lattice models for folding heteropolymers. Phys. Rev. 2005, E71:031906 |
| [20] | Kim SY, Lee S B, Lee J: Structure optimization by conformational space annealing in an off-lattice protein model. Phys. Rev. 2005, E72:011916 |
| [21] | Chen M, Huang W: Simulated Annealing Algorithm for Protein Folding Problem. Mini-Micro Systems 2007, 28(1):75-78. |
| [22] | Cheng W: Protein 3D Structure Prediction by Improved Tabu Search. Master dissertation of Wuhan University of Science and Technlogy 2009 |
| [23] | Mount DW: Bioinformatics: sequence and genome analysis. Cold Spring Harbor, Cold Spring Harbor Laboratory Press 2001 |
| [24] | Yang Q M, Yang Jack Y: Lecture notes: 2010 and beyond, the decade of high-performance computing for the next- generation sequence analysis. I. J. Computational Biology and Drug Design 2009, 2(2):204-206 |
| [25] | A. Hedar, A.F. and T. Hassan ,"Gentic Algorithm and Tabu Searched Methods for Molecular 3D-Structure Predicition", Numerical Algebra Control and Optimization, Vol. 1, No. 1, pp. 191–209, 2011 |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTML