-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Life Sciences
p-ISSN: 2163-1387 e-ISSN: 2163-1395
2017; 7(1): 5-10
doi:10.5923/j.als.20170701.02

Enhancing Soluble Gene Expression of α-amylase Bacillus licheniformis and Purification of Recombinant Protein
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAyesha Pervaiz1, Barizah Malik1, Naeem Rashid2
1Institute of Biochemistry and Biotechnology, University of the Punjab, Lahore, Pakistan
2School of Biological Sciences, University of the Punjab, Lahore, Pakistan
Correspondence to: Barizah Malik, Institute of Biochemistry and Biotechnology, University of the Punjab, Lahore, Pakistan.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

α-amylase is an endoglucan hydrolase that catalyzes the cleavage of α(1-4) glycosidic linkage between glucose residues of polysaccharides. α-amylase involved in starch degradation plays a wide role in the starch industry. Also, it has many applications in other industries as it is being employed in detergent industry, food industry, paper industry, pharmaceuticals and sugar industry. This enzyme shows optimized activity at 40-100°C and a pH of 6.5. α-amylase is a metalloenzyme and Ca+2 ions play an important role in its thermostability and activity. Recombinant α-amylase, from Bacillus licheniformis strain ATCC 27811, was produced in Escherichia coli. Enzyme expression was optimized and intracellular protein expression in soluble form was found with 306 total units of enzyme activity. The enzyme was heat treated at 70°C and it showed its thermostability. The enzyme was purified using the single step purification technique of Ni+2 affinity column chromatography.
Keywords: Bacillus licheniformis, Amylase, Expression, Nickel affinity chromatography, Starch plate assay
Cite this paper: Ayesha Pervaiz, Barizah Malik, Naeem Rashid, Enhancing Soluble Gene Expression of α-amylase Bacillus licheniformis and Purification of Recombinant Protein, Advances in Life Sciences, Vol. 7 No. 1, 2017, pp. 5-10. doi: 10.5923/j.als.20170701.02.
Article Outline
1. Introduction
- Starch is a widely distributed polysaccharide. Starch containing substrates are present in plants and some algae. It is composed of glucose subunits that are bound to each other through glycosidic linkages. It is made up of two subunits amylose and amylopectin [1, 2]. The soluble linear component of starch is amylose that forms α(1-4) glycosidic bond whereas branched polymer of starch amylopectin forms α(1-6) glycosidic bond [3, 4]. A lot of enzymes act on starch and degrade it. They are wholly classified into four major groups; exoamylases, endoamylases, debranching enzymes and transferases [5]. α-amylase belongs to the class of enzymes called endoglucanhydrolases that are involved in cleavage of α(1-4) glycosidic linkages in many polysaccharides including starch, glycogen and other oligosaccharides that are composed solely of glucose residues [6]. α-amylase is considered to be an extracellular enzyme but intracellular α-amylase is also reported that has unique characteristics as compared to extracellular α-amylase [7]. α-amylase has many applications on an industrial scale like paper industry, detergent industry, pharmaceuticals, and sugar industry but its foremost demand is in a starch industry where it is used for starch liquefaction [8-10]. This is the reason why α-amylase constitutes about 30% of industrial enzymes produced [6]. Bacillus licheniformis and Bacillus amyloliquefaciens are the bacillus species most frequently used for overproduction of α-amylase because they produce thermostable enzyme [11]. Cloning and expression of α-amylase are being practiced all over the world. Different expression vectors can be used for this purpose. After cloning the desired gene is expressed in the host strain by incubating in the presence of an inducer [12]. The expressed protein can then be purified using certain techniques that include ammonium sulfate precipitation, ultrafiltration, dialysis, ion exchange chromatography, column chromatography etc. These techniques require stepwise purification [13, 14]. Metal affinity chromatography is a single step purification technique. Expression vectors contain an additional sequence of 22 base pairs that code for six histidine residues. This histidine tag facilitates purification by metal affinity chromatography [14]. Nickel affinity column chromatography purifies up to 100-folds in a single step and gives 95% purity in high yield [12]. Expression of proteins in E. coli system often results in the formation of inclusion bodies that are inactive insoluble aggregates. [15-17].Therefore an approach to produce recombinant α-amylase in E. coli expression system in the soluble fraction using recombinant DNA technology was used.
2. Materials and Methodology
2.1. Bacterial Strains, Culture Conditions and Plasmids
- In this study Bacillus licheniformis ATCC 27811 was used as a source of the α-amylase gene. Escherichia coli DH5α was used for gene cloning, and E. coli BL21-CodonPlus (DE3)-RIPL was used for expression of the gene. All the three strains were grown in LB medium (1% tryptone, 0.5% yeast extract and 1% NaCl) at 37°C, and glycerol stocks were stored at –80°C. Plasmid pET-28a(+) (Fermentas Inc., USA) was used for the expression of the gene.
2.2. Cloning of N-amy Gene
- In a study conducted in our laboratory on α-amylase pET-21a(+) was used as an expression vector [18, 19]. N-amy gene coding for α-amylase from B. licheniformis strain ATCC 27811 was recovered from recombinant pET-21a(+)-N-amy by double digestion using restriction endonucleases HindIII and NdeI. The recombinant pET-21a(+)-N-amy vector was cleaved into two fragments. The fragment of size 1500bp encoding the N-amy gene was recovered using a Thermo scientific Gel Extraction. pET-28a(+) was also double digested by using same resctriction endonucleases HindIII and NdeI to produce complementary ends and larger fragment was recovered from the gel. The NdeI-HindIII restriction fragment from pET-21a(+)-N-amy was ligated into the pET-28a(+) expression vector at the corresponding sites. The resulting plasmid, pET-28a(+)-N-amy, was used to transform E. coli strain DH5α by following the chemical method of competent cell preparation [20]. The transformants were screened by restriction analysis. The expression vector pET-28a(+)-N-amy was extracted from the transformed cell culture using a Thermo scientific Miniprep Kit, double digested using same restriction endonucleases and visualized by using 1% agarose gel electrophoresis (1% agarose gel was prepared with 2μl of ethidium bromide [0.5μg/ml], 5µl of DNA sample was mixed with 2µl of 6X gel loading dye [10mM tris Cl buffer, pH 7.6, 30% glycerol, 10mM EDTA, 0.005% w/v bromophenol blue, 0.005% w/v xylene cyanol] along with DNA ladder [0.1µg/µl, Gene ruler, SM0311, 1Kb, Fermentas Inc], voltage of about 80 volts was applied to the system and visualized under the UV light). The recombinant plasmid pET-28a(+)-N-amy was then transformed into E. coli strain BL21-codon plus (DE3)-RIPL for the expression of the gene.
2.3. Expression of the α-amylase Gene
- Host cells containing the pET-28a(+)-N-amy plasmid were grown overnight at 37°C in LB medium containing kanamycin (30µg/ml). The overnight grew culture was diluted (1%) into fresh LB medium (1liter) containing kanamycin (30µg/ml), and was incubated at 37°C until A660 reached 0.4-0.6. Heterologous expression of the α-amylase gene was induced by the addition of 0.1, 0.5 or 1mM (final concentration) isopropyl-β-D-thiogalactopyranoside (IPTG), and expression was optimized at different temperature conditions, time of incubation and IPTG concentrations.
2.4. Electrophoretic Analysis
- Cell pellet was sonicated by resuspending cell pellet in 20mM tris Cl buffer (pH 8.0). The expression level of the recombinant α-amylase was determined by 15% SDS-PAGE (voltage of about 100 volts was applied to the system) [13] followed by staining with Coomassie Brilliant Blue and destaining with a solution of methanol-glacial acetic acid–water (5% methanol, 7% acetic acid and 88% H2O).
2.5. Enzyme Activity Assays
- Enzyme assays were performed to study the activity of enzyme. Amylose in starch gives blue color in the presence of iodine, reduction in the blue color indicate degradation of starch by α-amylase. To check the activity of α-amylase quantitative iodine assay was performed in which 50μl of enzyme solution was incubated at 80°C for 15min with 90μl of 1% soluble starch solution, the reaction was terminated by quenching on ice for at least 10 min and then centrifuged for 3-4 min. 60μl of the reaction mixture was removed and mixed with 100μl of 0.1N NaOH. This solution (100μl) was mixed with 2ml of freshly prepared iodine solution and absorbance was measured at 660nm. Total units of α-amylase were calculated by following formula:Total Units = Δ OD X Dilution Factor X Total volume of enzyme / Volume of enzyme in reaction
2.6. Plate Assay
- Following the similar principle, qualitative starch plate assay was also performed. For this 1% starch agar plate was made and 10μl of the enzyme solution was spotted on the plate and left in an incubator at 70°C for one hour. After one hour it was stained with potassium iodide solution (0.005% [wt/vol] I2 and 0.01% [wt/vol] KI). Hollow circles appeared on the plate where the enzyme was active.
2.7. DNS Assay
- DNS gives a yellow colored product with reducing sugars. In assay 100µl of diluted enzyme solution was added to 200µl of 0.1mol/liter sodium acetate (pH: 5.0) and 200µl of 1% (w/v) soluble starch, and incubated for 20 min at 80°C. The reaction was stopped by quenching on ice, then 500µl of 1% DNS was added and boiled for 10 min. Absorbance was measured at 540nm after cooling to room temperature. Control experiments included a reaction mixture without substrate.
2.8. Heat Treatment of Crude Extract
- To check the thermostability of soluble and insoluble fractions, samples were heat treated at 70°C for 15 min. The samples were analyzed by 15% SDS PAGE gel electrophoresis.
2.9. Purification of α-amylase
- The recombinant α-amylase in the soluble fraction was precipitated with 85% ammonium sulfate and dialyzed against 20mM tris Cl, pH 8.0. Polyhistidine-tagged α-amylase was purified from other bacterial proteins present in soluble fraction according to the purification procedure at native conditions from Invitrogen manual of Ni+2-NTA purification system. Imidazole concenteration up to 500mM was used for elution of polyhistidine-tagged recombinant protein. Eluted fractions were dialysed against 20mM tris Cl pH 8.0 for 3 hour and precipitated using acetone (1 volume of sample was mixed with 2 volumes of ice-chilled acetone and kept at -20°C overnight, the sample was centrifuged later at 4000rpm for 30 min, pellet was washed with 70% ethanol and again centrifuged and ethanol was allowed to evaporate completely). Precipitates of eluted fractions were resuspended in 20mM tris Cl pH 8.0 and were assayed for the presence of eluted polyhistidine-tagged α-amylase by analyzing 20µl aliquots by electrophoresis through a 15% SDS-polyacrylamide gel.
2.10. Estimation of Enzyme by Bradford Assay
- Total protein concentration in the purified product of α-amylase was estimated by the dye binding assay using bovine serum albumin as a standard.
3. Results
3.1. Cloning of N-amy
- Recombinant plasmid pET-28a(+)-N-amy (having HindIII and NdeI sites) was isolated and was digested by restriction enzymes HindIII and NdeI. Restricted band of N-amy gene was purified from gel and then analyzed by 1% agaose gel (Figure 1).
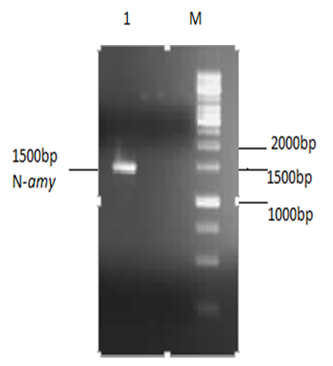 | Figure 1. 1% Agarose gel analysis of purified restricted N-amy: Lane M shows gene ruler 1Kb DNA ladder (Fermentas) and Lane 1 shows purified N-amy gene of 1500bp |
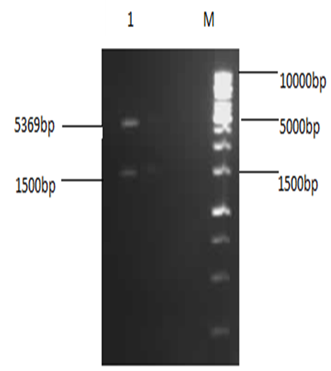 | Figure 2. 1% Agarose analysis of restricted plasmid: Lane M shows 1Kb DNA ladder (Fermentas) and lane 1 shows double digested pET-28a(+) |
3.2. Expression of α-amylase
- The E. coli strain BL21- Codon plus (DE3)-RIPL transformants obtained after cloning were used for expression studies. The N-amy was expressed in E. coli strain BL21- Codon plus (DE3)-RIPL by induction with IPTG. In a study conducted in our laboratory, N-amy gene was cloned in pET-21a(+) [19]. In this expression vector maximum expression of α-amylase (in soluble fraction) was obtained at 0.01mM IPTG concentrations and at temperature 18°C. So to study the expression of α-amylase in pET-28a(+) conditions like IPTG concentrations and temperature were optimized. IPTG concentrations of 0.1mM (Figure 3), 0.5mM (Figure 4) and 1mM IPTG (Figure 5) were used at varying time intervals (4-8 hours incubation) and temperature conditions (18°C and 37°C). Cell pellet was sonicated in the presence of 20mM tris Cl pH 8.0. Maximum expression of recombinant protein was observed after induction with 1mM IPTG for 4 hours at 37°C in the soluble fraction (Figure 5). Expression was analyzed by 15% SDS-PAGE.
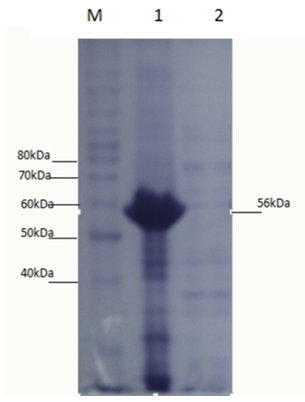 | Figure 3. 15% SDS gel of 0.1mM IPTG induction: Lane M shows protein ladder (Invitrogen), lane 1 shows an insoluble fraction of cell lysate after sonication and lane 2 shows a soluble fraction of cell lysate after sonication |
 | Figure 4. 15% SDS PAGE analysis of 0.5mM IPTG induction: Lane M is protein ladder (Invitrogen), Lane 1 is an insoluble fraction of cell lysate obtained after sonication and lane 2 is a soluble fraction of cell lysate after sonication |
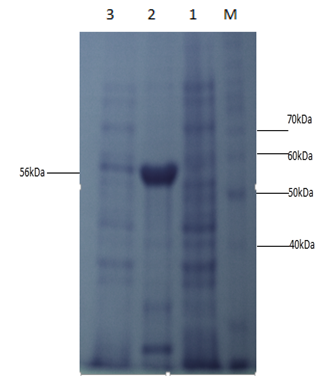 | Figure 5. 15% SDS PAGE analysis of 1mM IPTG 4h incubation: Lane M shows protein ladder (Invitrogen), lane 1 shows uninduced cell lysate, lane 2 shows a soluble fraction of 1mM IPTG after sonication and lane 3 shows an insoluble fraction |
3.3. Enzyme Activity Assay
- Iodine assay of soluble and insoluble fractions was performed in duplicates. The following table 1 shows the absorbance at 660nm indicating the activity of cell lysates.
|
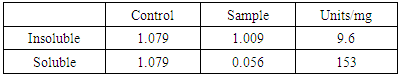
3.4. Starch Plate Assay
- Starch plate assay of crude extract was performed to check the activity of soluble and insoluble fractions of 1mM IPTG induction. Formation of a clear zone indicated the activity of α-amylase to degrade 1% starch solution (Figure 6).
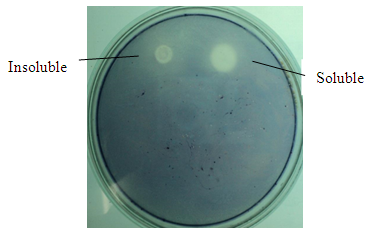 | Figure 6. Starch plate assay of soluble and insoluble fractions |
3.5. DNS Assay
- DNS assay was also performed for further analysis of activity of the soluble and insoluble fractions. Similar results were obtained showing soluble fraction to be active while insoluble one to be only slightly active.
3.6. Heat Treatment of 1mM IPTG Cell Lysate
- To check the thermostability of α-amylase, cell lysates were incubated at 70°C for 15 min then 15% SDS-PAGE analysis was performed to verify. Results from the gel showed enzyme stability at higher temperatures.
3.7. Purification of α-amylase from Ni+2 Affinity Chromatography
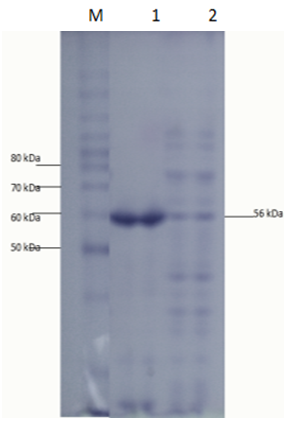
- α-amylase was purified by Ni+2 affinity chromatography and then the purified product was verified by 15% SDS PAGE gel electrophoresis. Single band formation indicated the purification of α-amylase (Figure 7).
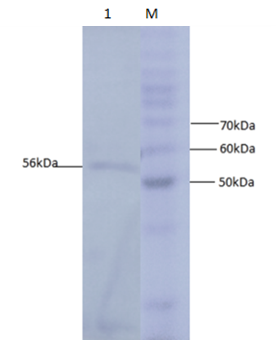 | Figure 7. 15% SDS PAGE analysis of purified protein Lane M is the protein ladder (Invitrogen) and lane 1 indicates the single band of purified α-amylase |
4. Conclusions
- Starch is being produced in a large number of economically important crops like wheat, rice, maize, potato, and tapioca. A large-scale starch processing industry has evolved in the last century. In the past decades, acid hydrolysis of starch has been replaced with the use of starch-converting enzymes producing maltodextrin, modified starches, or glucose and fructose syrups. Nowadays, these enzymes constitute about 30% of the world's industrial enzyme production. These enzymes are not only used in starch hydrolysis, but also in a number of other industrial applications, such as laundry and porcelain detergents or as anti-staling agents in baking. A number of these starch-converting enzymes belong to a single family: the α-amylase family. Because of so much importance of α-amylase in industries purified products of this enzyme are being produced. It would be highly advantageous to have access to large quantities of pure amylase to allow studies on its structure and overproduction. Recombinant DNA technology would aid in the large-scale production of α-amylase. Therefore, the aim of the present study is to produce recombinant α-amylase in the soluble fraction in its active form and purify it using Ni+2 chromatography.E. coli strain BL21-CodonPlus (DE3)-RIPL expression system and pET-28a(+) as expression vector were used for expression of N-amy gene. Gene was cloned in pET-28a(+) vector under the control of the inducible T7 promoter and His-tag fusion partner and pET-28a(+). N-amy was expressed in bacterial strain BL21-CodonPlus (DE3)-RIPL. Expression was optimized to gain maximum yield of the enzyme in E. coli expression system. IPTG induction was carried out at 37°C and concentration of 0.1mM, 0.5mM and 1mM for different time intervals ranging from, 4 hours and 8 hours in LB. The maximum expression was obtained after 4 hours of post induction at 37°C and IPTG concentration of 1mM in LB medium. A band of recombinant α-amylase was observed at approximately 56kDa after sonication of the soluble fraction. Quantitative and qualitative starch assays were performed to check the activity of soluble and insoluble fractions. Results showed the maximum activity of the enzyme in soluble fractions. Total units/mg of α-amylase was calculated to be 153. Recombinant α-amylase was thermostable up to 70°C. The enzyme was purified by ammonium sulfate precipitation followed by Ni+2 affinity column chromatography. Using Bradford assay enzyme concentration was estimated to be 310μg/ml from the standard curve.As this study gives a way to produce α-amylase in the soluble fraction without encountering problems associated with misfolding and inclusion bodies. So purified products of the soluble and active enzyme can be obtained following this protocol. Moreover, the enzyme was also thermostable at 70°C. Since starch is only soluble at temperatures close to 100°C, one key in its industrial hydrolysis must be the use of highly thermostable enzymes. So enzyme produced has a high applicability in starch industry.