-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Life Sciences
p-ISSN: 2163-1387 e-ISSN: 2163-1395
2013; 3(2): 28-34
doi:10.5923/j.als.20130302.02
Creation of Sugar Transporter Gene (uspC) Knockout Constructs in Mycobacteria
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLStephen Kugbere Agadagba
Department of Biochemistry, Faculty of Life Sciences, University of Benin, Benin City, Nigeria
Correspondence to: Stephen Kugbere Agadagba, Department of Biochemistry, Faculty of Life Sciences, University of Benin, Benin City, Nigeria.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The processes involved in the selection and uptake of nutrients in Mycobacteriumtuberculosis are not fully understood. Specifically, the roles of the transport proteins and theidentities of their substrates are very crucial in gaining a clearer understanding of the pathogen. In the current study, uspC gene knockout constructs were successfully created and extracted from transformed top 10 E.colicells. The process involved PCR amplification of the left flank (LF) and the right flank (RF) of the UspCgenehomolog (Msmeg4468) in M. smegmatis, followed by ligation of AlwnI-digested PCR fragments and van91I-digested plasmid (p0004S) to generate the UspC knockout constructs. Confirmation of the UspC knockout constructs was done by visualizing the digested fragments in an agarose gel electrophoresis after digestion with PacI restriction endonuclease. The results revealed that the transformed top 10 E.coli cells contained the uspC gene knockout constructs of size 8kb. Although the UspCknockout constructs were not used to create genetic knockout mutants in Mtb and M.smegmatis due to time constrains of the research, however the present study can serve as a foundation for future scientific research to understand the specific roles of the UspC sugar transporter in the Mycobacteria family.
Keywords: Mycobacterium tuberculosis, Mycobacterium smegmatis, Sugar transporters, UspC (Msmeg4468), Knockout constructs, Polymerase chain reaction
Cite this paper: Stephen Kugbere Agadagba, Creation of Sugar Transporter Gene (uspC) Knockout Constructs in Mycobacteria, Advances in Life Sciences, Vol. 3 No. 2, 2013, pp. 28-34. doi: 10.5923/j.als.20130302.02.
Article Outline
1. Introduction
- Tuberculosis (TB) is an infectious and virulent disease commonly caused by Mycobacterium tuberculosis (Mtb) in humans. TB is a major cause of mortality, globally killing over 5,000 patients daily and thus accounting for approximately 2 million deaths annually[1]. A healthy individual may initially be infected with Mtb without exhibiting any symptoms because the immune system provides protection against the bacteria. However, with reduction in the defence mechanism of the individual’s immune system, Mtb becomes fully active and exhibits virulence. Despite recorded success in the exploitation of genetic techniques for studying mycobacteria, the identity of all the carbon sources and nutrient transport proteins in the pathogen are not completely known[2]. However, for those transport proteins that have been identified and characterised, many have been experimentally proven to function in the acquisition of small molecules including sulphates, phosphate and aminoacids, as well as other essential nutrients such as lipids (particularly cholesterol)[3,4,5,6]. A recent scientific study suggests that in contrast to lipids, the Mtb pathogen can have great limitation in obtaining sugars from the host’ cells[7]. This is true because gluconeogenesis has been implicated to be an indispensable pathway for attaining virulence by Mtb[7]. Furthermore, sugar acquisition has been reported to be a major necessity for the survival of Mtb, especially during the second stage (non-dormant phase) of tuberculosis infection[2].M. smegmatis, a model organism of Mtb is able to grow on several sugar and carbohydrate sources such as pentoses, hexoses and polyols[8] It has been previously established from in-silico analysis of mycobacteria genome that M. smegmatis possesses 28 carbohydrate transporters in its inner membrane[9]. More than one-half of the sugar transporters in M. smegmatis come from a broad family of transporter proteins called the ATP-binding cassette (ABC)[10]. ABC-transporters are transmembrane proteins that hydrolyse cellular energy (ATP) to transport various substances such as sugars, lipids, peptides, aminoacids, drugs, etc, across the cell membrane[11]. These transporters generally have two binding domains namely: a nucleotide binding domain (NBD) located in the cytoplasm, and a transmembrane domain (TMD) that spans the lipid bilayer[12]. Besides the ABC transporters, M. smegmatis also has a few other transport proteins belonging to specific groups namely the sodium solute superfamily (SSS), the major facilitator superfamily (MFS), the major intrinsic protein (MIP) family and phosphotransferase system (PTS)[2]. Transport proteins of these families are separately involved in the acquisition of galactose, glucose, glycerol and fructose across the inner membrane. This therefore implies that M. smegmatis has the appropriate machinery for transporting the sugars mentioned above. In contrast, Mtb only has six sugar transporters of which four are ABC transporters and two belong to the MFS. These transporters have been previously discussed in earlier research works[12, 13]. However, a brief description of the ABC transporter (uspC) that is relevant to the current study is as follows. The uspC acts as a periplasmic sugar binding lipoprotein of the ABC transporters[7] that is closely associated with uspB and uspA inner membrane transport proteins. Although, uspC is noted to be a sugar transporter protein, however its specific sugar substrate remains unknown. The uspABC genes (loci: Msmeg4466 - Msmeg4468) in M. smegmatis are homologous to the uspABC genes (loci: Rv2316 - Rv2318) in Mtb[7]. These similarities enable researchers make appropriate comparisons between both mycobacteria species and draw rational conclusions about previously unknown facts in the physiology and cellular metabolism of Mtb. A clear understanding of the cellular metabolism in Mtb is extremely important in discovering new strategies to combat TB infection. Current drug therapies are of great value in reducing spread of TB, but are still quite limited especially for treating drug resistant forms of the disease. Consequently, a strategy exploited in scientific research to identify new anti-TB drug targets involves studying the roles of specific transport proteins in Mtb. In this regard, techniques that are frequently employed include gene knock out systems and site-directed mutagenesis that generate null mutants, whose phenotypes can be examined. As stated above, sugar acquisition is paramount for Mtb propagation in certain stages of TB infection, thus knock out of specific genes (for example the uspC gene) encoding sugar transport proteins may attenuate the pathogen’s virulence. Thus, the aim of the current research is to make knockout constructs for the uspC gene and possibly create genetic mutants of uspC in M. smegmatis and M. tuberculosis, and observe the resulting phenotypes.
2. Materials and Methods
2.1. Materials
- All reagents and chemicals used in this research were bought from Merck and Sigma-Aldrich, unless specifically stated. All the enzymes used were obtained from New England Biolabs (NEB).
2.2. Bacterial Strains
- Top 10 Escherichia coli strains were used in this study. Competent stocks of the strains were made up in 2.5 mL calcium chloride (100 mM) and 15% glycerol mixture, and stored away at -80ºC. When required, top 10 E. coli cells were used for transformation experiments involving creation of knockout mutants in M. smegmatis.
2.3. Extraction and Digestion of Aprar p0004S Cosmids from Hygromycin Resistant top 10 E. coli Cells
- Top 10 cells containing apraR p0004S cosmids were grown in shake flasks for 24 h in Luria-Bertani (LB) broth at 37ºC and 180 rpm. The apraR p0004S cosmid contained the hygromycinr-sacB cassette. The media was supplemented with Hygromycin (hyg) to a final concentration of 150 μg/ml, to select viable cells. After growth, the cosmids were extracted from the cells by following the manufacturer’s instructions (QIAGEN, 2006) in a QIAprep spin mini-prep kit. The concentration of the extracted plasmids, were measured via a Nanodrop 2000/2000C program (thermal scientific). About 50 μL of the extracted cosmids were digested for 1h by Van91I restriction endonuclease. Each sample (10 μL) of the digested cosmids was separated by agarose gel-electrophoresis (1% agarose gel), for 1 h at 140 Volts (V) and 400 milliAmperes (mA). The separated bands were visualized in UV-light and the fragments of interest were excised from the gel with a scalpel and the total weight of the extracted fragments was recorded. Excised fragments were resuspended in 3×QG buffer and 1mL aliquots were stored at -20ºC, until required.
2.4. Polymerase Chain Reaction Amplification of Gene Regions Adjacent to the UspC gene homolog in Mycobacteria smegmatis
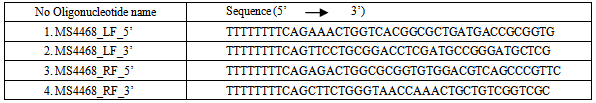
- The left flank (LF) and the right flank (RF) of the UspC gene homolog (Msmeg4468) in M. smegmatis, were amplified by polymerase chain reaction (PCR) in a T3 thermocycler (Biometra®). Specifically, for the uspC gene knockout (K.O) system, the 68LF and 68RF were amplified. The reaction vessel for the PCR contained: the genomic DNA template of M. smegmatis (0.5 μL), primers (for LFs and RFs of the specified regions), dimethylsulphoxide (DMSO), Pfu DNA polymerase enzyme, 10×Pfu buffer, deoxynucleoside triphosphates (dNTPs) and distilled water (H2O). The base sequences of the primers used are highlighted on the table below.
|
2.5. Purification and Digestion of the PCR Amplified Products
- The PCR amplified flanks (68LF and 68RF) were purified by following the manufacturer’s instructions (QIAGEN, 2006) in a QIAquick PCR purification kit. The concentrations of the pure PCR amplified flanks were recorded in the Nanodrop as stated in section 2.4 above. Afterwards, 25 μL of the 68LF and 68RF respectively, were digested by the restriction enzyme AlwnI. This was done to create sticky ends in the amplified and purified flanks.
2.6. Ligation of AlwnI-digested Flanks and the Van 91I-digested Plasmid (p0004S) Togenerate the uspC Gene K.O Ligated Constructs (K.O Plasmids)
- AlwnI-digested flanks (68LF, 68RF) for the uspC gene were mixed in two separate epidoff tubes (1.5 mL) containing thawed Van91I-digested p0004S plasmid. Subsequently, the mixtures were separately eluted by following the manufacturer’s instructions (QIAGEN, 2006) in a QIAquick gel extraction kit. T4 buffer (2.5 μL) and DNA ligase enzyme (1.5 μL) were both added to the uspC gene K.O eluents, to trigger the ligation reactions for generating their respective K.O plasmids. The ligation reactions were allowed to continue overnight at 4ºC.
2.7. Transformation of Competent top10 E. coli cells with the uspC K.O Constructs (K.O plasmids)
- Aliquots (5 μL respectively) of the uspC K.O construct were used to transform 50 μL competent top 10 E. coli cells in two round-bottom falcon tubes (15mL). Both tube mixtures were left stationary for 30 min at 4ºC and subsequently heat shocked in a dry water-bath (LABNET) for 30 sec at 42ºC. This step was done to enable the cells to take up the ligated constructs or K.O construct. The mixtures were left stationary for 2 min at 4ºC, then 200 μL LB broth was aseptically added to both tubes and the cultures were incubated with shaking for 1h at 37ºC and180 rpm. After 1 hour, the grown cultures (50 μL each) were spread with glass beads on hygromycin-B (150 μg/mL) supplemented LB agar plates. The spread plates were incubated overnight at 37ºC. Single grown colonies from the overnight agar plates were picked with pipette tips (20 μL) and aseptically inoculated in 4 Ml LB broth supplemented with hyg B (150 μg/mL). The cultures were incubated overnight, with shaking at 37ºC and 180 rpm.
2.8. Extraction, Digestion and Sequencing of the uspC K.O Construct from Transformed Top 10 E. coli Cells
- The overnight grown cultures of transformed top 10 cells were harvested by centrifuging at 4,000rpm for 10 min. The supernatants were discarded and the cell pellets were used to isolate the K.O plasmids. Extraction of the uspC gene K.O plasmids was done as previously stated in section 2.4 above. The concentrations of the isolated uspC K.O plasmids were measured on the Nanodrop. Two groups (5 μL each) of the plasmids were setup and separately digested with Van91I and PacI restriction enzymes for 1h respectively. The digests were subjected to agarose gel electrophoresis, as previously stated above. This was done to initially confirm the extracted uspC gene K.O constructs.
2.9. Creation of uspC (Msmeg4468 gene) Knockout in M. smegmatis
- Due to the short period of time given for this study (3 months), the sequenced uspC gene ligation construct could not be used to knockout the UspC gene (Msmeg4468) in M. smegmatis and observe the resulting phenotypes.
3. Results
3.1. ApraR p0004S Cosmid Extraction and Digestion
- ApraR p0004S cosmids were extracted from grown cultures of top 10 E. coli by following a miniprep protocol (QIAGEN, 2006). The presence of the Hygr-SacB cassette in apraR p0004S enabled the top 10 cells to grow in the Hyg (150 μg/mL)-supplemented LB broth. The extracted cosmids were digested by Van91I restriction endonuclease to obtain a mixture of fragments which were separated on an agarose gel (figure 1). The required fragments (3.6 kb and 1.7 kb) were visualized in UV-light and successfully excised from the agarose gel for further use. The total weight of the excised fragments was 14 g.
3.2. PCR Amplification of the Flanking Regions around uspC (Msmeg4468), Purification and Digestion of the PCR Products
- The left flank (68LF) and right flank (68RF) of uspC were amplified by PCR with primers specific to the regions of interest. Small samples (10 μL) of the PCR products were run on an agarose gel to separate the fragments. From the agarose gel results (figure 2), thick bands (1kb) of the PCR products were observed, which indicated that the flanking regions of uspC gene (Msmeg4468) in M. smegmatis, had been successfully amplified. The amplified flanks were subjected to purification by following the manufacturer’s instructions (QIAGEN, 2006) in a QIAquick PCR purification kit. Afterwards the concentration of the purified flanks were measured and recorded as follows: 68LF; 4.5 ng/μL and 68RF; 3.3 ng/μL. The purified flanks were also digested with AlwnI restriction endonuclease and this yielded sticky ends that were suitable for the ligation reaction.
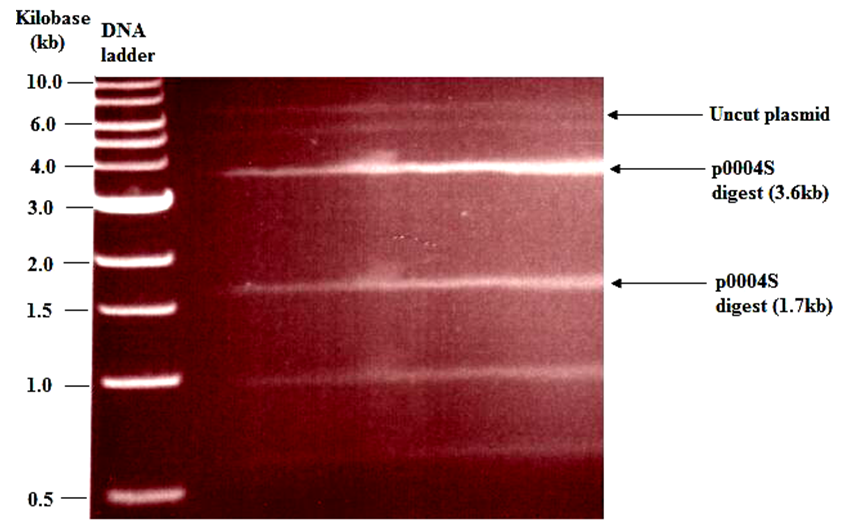 | Figure 1. Agarose gel electrophoresis showing: fragments of Van91I digested apraR p0004S cosmid. The topmost indistinct band represents uncut plasmid or super-coiled plasmid. The sizes of the vector fragments of interest were 3.6kb and 1.7kb |
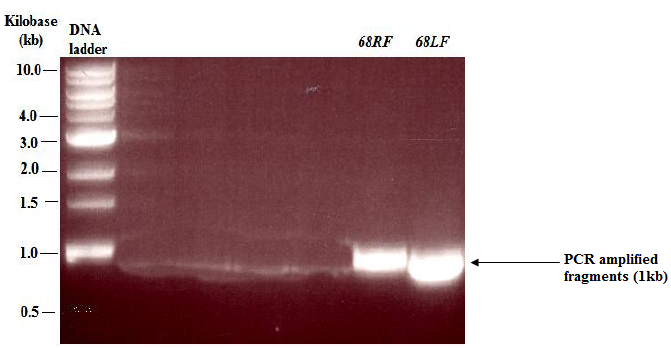 | Figure 2. Agarose gel electrophoresis showing: PCR amplified flanks of M.smeg4468 gene from M. smegmatis Mc2. RF: right flank; LF: left flank. The size of each flank was 1kb |
3.3. Transformations with the K.O Constructs
- AlwnI PCR digests were mixed with Van91I-digested p0004S in the presence of DNA ligase to generate the uspC gene K.O constructs (ligated plasmid constructs). These constructs were separately used to transform 50 μL competent top 10 E. coli cells that were then grown for 1h in LB media. After incubation, the cultures appeared cloudy, which indicated cell growth in all the cultures containing the uspC gene K.O constructs. These cultures were spread on LB agar plates (supplemented with hyg) and incubated at 37ºC to screen for colonies that were successfully transformed and thus resistant to hygromycin antibiotic. From the results, visible colonies were observed in agar plates transformed with the uspC gene K.O constructs.
3.4. Extraction and Digestion of uspC Gene K.O Constructs
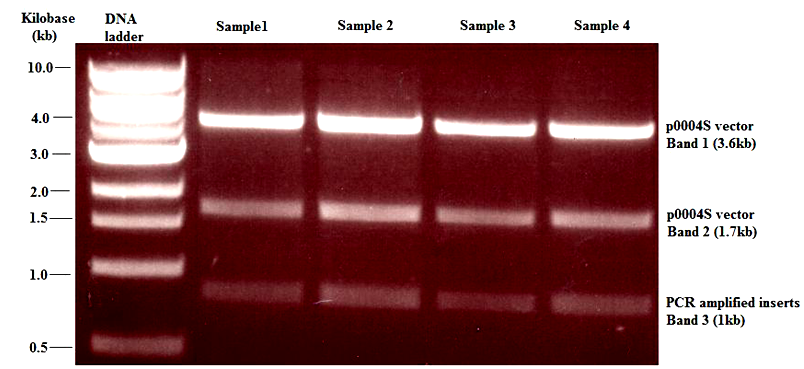 | Figure 3. Agarose gel electrophoresis showing: banding pattern obtained from the uspC gene (Msmeg4468) K.O construct after digestion with Van91I restriction endonuclease. The first two bands contained Van91I-digested fragments of p0004S vector. Band 3 was suspected to be an overlap of two 1kb fragments containing the PCR amplified inserts of uspC gene digested with AlwnI endonuclease. This was confirmed by was by digestion of the K.O construct with PacI endonuclease (see figure 4) |
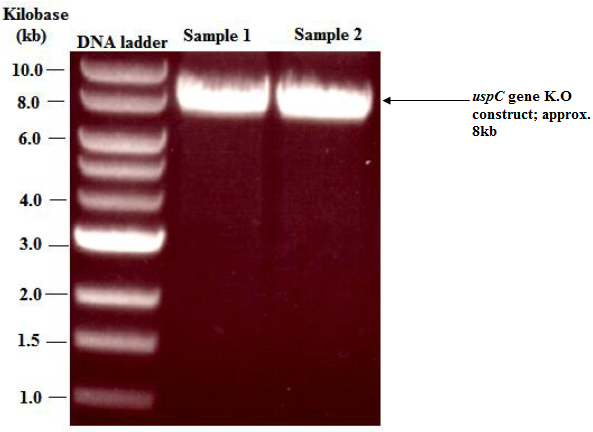 | Figure 4. Agarose gel electrophoresis showing: banding pattern obtained from the uspC gene (Msmeg4468) K.O construct after digestion with PacI restriction endonuclease. Digestion of the uspC knockout construct with PacI restriction endonuclease, linearised the vector and showed that the total size of the construct was approximately 8kb |
- Top 10 colonies that were successfully transformed with the uspC gene K.O construct were selected and used to generate an overnight culture in Hyg (150 μg/ml)-supplemented LB broth. As expected the colonies were resistant to hyg and produced a dense cell culture that was used to extract uspC gene K.O constructs (ligated constructs) in a miniprep procedure (QIAGEN, 2006). The concentration of the extracted constructs ranged from 74 - 247 ng/ μl. Aliquots of the ligated construct for the uspC gene K.O were separately digested with restriction endonuclease Van91I and the fragments were separated on agarose gels. Digestion of the ligated construct with Van91I ideally should produce four distinct bands of sizes 3.6 kb (representing the longer fragment of Van91I-digested p0004S cosmid), 1.7 kb (representing the shorter fragment of Van91I-digested p0004S cosmid), 1 kb (representing the first flanking region of uspC) and 1 kb (representing the second flanking region of uspC). However, in this study only the first three bands (3.6, 1.7 and 1 kb) were observed (figure 3). Due to the thickness of the third band, it was suspected to be an overlap of two 1kb fragments. Subsequently this was confirmed by digestion of the ligated construct with PacI restriction endonuclease, which linearised the vector construct and showed that the total size of the ligated construct was approximately 8 kb (figure 4).
4. Discussion
- A gene knockout is a molecular biology technique in which one of an organism's genes are made inoperative or non-functional[14]. Knockouts are used in learning about a gene that has been sequenced, but which has an unknown or incompletely known function. This implies that knockouts enable scientists to gain a primary understanding of the role of a specific gene or DNA region by comparing the knockout organism to a wild-type with a similar genetic background. In gene knockout systems, the K.O construct is engineered to recombine with the target gene, which is accomplished by incorporating sequences from the gene itself into the construct[3]. Recombination then occurs in the region of that sequence within the gene, resulting in the insertion of a foreign sequence to disrupt the gene[15]. With its sequence interrupted, the altered gene in most cases will be translated into a non-functional protein, if it is translated at all. Despite the wealth of genomic data and availability of sophisticated genetic tools, the identity of many transporters for essential nutrients of M. tuberculosis and other mycobacteria still remains unknown[2].
5. Conclusions
- From the foregoing, the current study attempted to understand the specific roles of the ‘so-called’ uspC sugar transporter by successfully creating K.O constructs of the uspC gene in Mycobacterium smegmatis. The knockout construct was engineered to recombine with the target uspC gene, which was accomplished by incorporating sequences from the uspC gene itself into the construct. Recombination was anticipated to occur in the region of that sequence within the uspC gene, resulting in the insertion of a foreign sequence to disrupt the uspC gene. Subsequently, the altered uspC gene in most cases will be translated into a non-functional protein, which may be unable to transport essential sugars across the membrane of Mycobacteria tuberculosis. This could reduce the virulence of the pathogen because it will be unable to use the hosts’ sugar reserves, which is required for M. tuberculosis metabolism and pathogenicity. The present study has successfully created the usp C knockout construct however, due to time constraints; the construct was not used to knockout the uspC gene. In this regard, further research is still on-going and a successful knockout of the uspC gene in M. tuberculosis is anticipated. This study therefore contributes greatly to the current knowledge base of M. tuberculosis research in the following ways. First, by creating (for the first time) a knockout construct of the uspC gene that encodes the uspC sugar transporter protein. Secondly, because the actual role of the uspC gene is not fully understood, creation of the uspC K.O construct in this study serves as a foundation for future research to clearly understand the roles of the gene, including the specific sugar substrates of the uspC protein it encodes. Thirdly, the current study provides an avenue for rational design and development of M. tuberculosis drugs that can specifically target the essential uspC sugar transporter protein, thus inhibiting sugar acquisition in M. tuberculosis and possibly attenuating the virulence of the pathogen.
ACKNOWLEDGEMENTS
- My appreciation goes to Prof. G.S. Besra and Dr. Apoorva Bhatt, for giving me the rare opportunity to work in the Mycobacteria lab, University of Birmingham, United Kingdom. I also wish to thank other members of the Mycobacterial lab including Dr. Sarah Batt and Albel for their kind assistance during this study. I am very grateful to my mum; Helen Agadagba and my siblings, Sharon, Rita, Edirin and Benjamin for their unfailing support and encouragement which always kept me strong. Finally, my utmost thanks go to God Almighty for His boundless love.
Conflict of Interests
- The author declares that he has no conflict of interest regarding the publication of his research article.