-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Life Sciences
p-ISSN: 2163-1387 e-ISSN: 2163-1395
2012; 2(6): 156-169
doi:10.5923/j.als.20120206.03
The Nature of Mildly Deleterious Mutations Eliminated upon Sexual Reproduction in Meiosis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLChubykin V. L.
Vavilov Institute of General Genetics, Russian Academy of Sciences; Gubkina 3, Moscow, 119991, Russia
Correspondence to: Chubykin V. L., Vavilov Institute of General Genetics, Russian Academy of Sciences; Gubkina 3, Moscow, 119991, Russia.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Mildly deleterious mutations (MDMs) with incomplete dominance, which decrease the viability of the progeny, apparently play a significant role in the evolution of sexual reproduction. In particular, they are thought to be eliminated in meiosis. The nature of MDM remains unclear. By studying the accumulated MDMs in Drosophila strains carrying various meiotic mutations (c(3)G17, mei-P22, mei-W68, mei-41, mei-218), we found that impair the formation of DNA breaks is more effective in accumulation of MDMs. The relationship between survival and generation number upon MDMs accumulation suggests that MDMs interaction corresponds to their synergistic epistasis. The viability in progeny after meiosis in heterozygotes with chromosome with accumulated MDMs and normal chromosome, and in heterozygotes with independently accumulated MDMs chromosomes was shown to be restored. Our results support the hypothesis that MDMs have epigenetic nature. It is proposed that: during the life cycle “mutant” variants of the formation of structural and functional loop domains appear in the chromosomes; these variants are normally corrected in meiosis; an abnormal loop alters the activity of many genes (~17), increasing (+) or decreasing (–) it. The hybrids with chromosomes carrying independently accumulated MDMs partially restore viability due to complementary interaction of + and – genes.
Keywords: Sex Evolution, Mildly (Slightly) Deleterious Mutations, Meiosis, Chromosome Loop Domains, Epigenetic Mutations, Drosophila Melanogaster
Cite this paper: Chubykin V. L., The Nature of Mildly Deleterious Mutations Eliminated upon Sexual Reproduction in Meiosis, Advances in Life Sciences, Vol. 2 No. 6, 2012, pp. 156-169. doi: 10.5923/j.als.20120206.03.
Article Outline
1. Introduction
- Diploidy, multicellular organization, the increase in the genome size and the structural complexity of organisms are associated with the development of sexual reproduction. The evolutionary advantage of sexual reproduction (which is in fact less effective and energetically less beneficial) over asexual one is not quite understood[1],[2]. More than 20 hypotheses have been advanced to explain this phenomenon[3]. In the evolution of sex, mildly deleterious mutations (MDMs), the effects of which are intermediate between neutral and deleterious, are thought to play a significant role. MDMs in homozygous state result in mortality of only part of the progeny. Moreover, they are semidominant and manifest in heterozygotes and these properties of MDMs provide their inheritance and maintenance in populations[1-5]. A high rate of appearance of new MDMs makes evolution with accumulation of advantageous mutations impossible[1]. Herman Muller was the first to theoretically demonstrate (later it was documented experimentally) that in asexual organisms, the gene pool is slowly but consistently degraded owing to the accumulation of MDMs (“Muller’s ratchet”)[4],[5]. The MDM number in such organisms increases with decreasing population size and increasing complexity of the genome (increasing number of genes). According to Muller, asexual populations, in spite of the mutation pressure, exist because of the simplicity of their organization (small genome size), extremely large population sizes, and strong stabilizing selection, rapidly eliminating MDM carriers, which are replaced by mutation-free clones. In this connection, an alternative explanation is that MDMs are eliminated in sexual reproduction[6],[7]. The mechanism of the elimination and its association with meiosis are unclear. Hypotheses highlighting the role of recombination (generation of MDM-free recombinant forms) in effective elimination of MDMs and, consequently, in reducing the mutational load of sexual populations[3],[5], hold only on unlikely condition of constant environmental changes, when in each generation new genotypes with high fitness would be required[1]. Indeed, the axiom on the evolutionary role of recombination has a serious defect, which lies in recombination itself. The high recombination rate, which is actually observed in nature, would destroy a beneficial gene combination not later than in the following generations. A simple reshuffling of not eliminated MDMs would have no effect on the progeny viability at the population level, which is the same proportion of the progeny as before recombination. In papers devoted to the study of MDMs, less discussed issue is their nature. Is MDM the "classical" mutations with changes in the nucleotide sequences of structural genes, or there are changes in the regulation of their activity? It is unknown. Incomplete dominance is used for classification of regulatory mutations and is associated with changes in the activity of structural genes[8]. Based on this MDM property, Mukai[9] suggested that MDMs are located in noncoding regions. Another suggestion connects reduce the viability of offspring with numerous insertions of mobile elements[10], which affect gene activity. Finally, it was concluded about the absence of MDMs upon mutagenic exposure[11]. The method of MDM accumulation in Drosophila, consisting in disturbance of main meiotic processes in an individual chromosome pair, confirms the role of meiosis in their elimination. A circuit method of MDM registration and accumulation in Drosophila was first proposed by Muller in 1928[12]. This method involves suppression of meiotic pairing and recombination in the examined chromosomes in many generations. With this aim, only heterozygotes with the corresponding chromosomes carrying multiple inversions and transpositions (balancers and crossover suppressors) were employed for reproduction. Chromosomes with multiple rearrangements are usually lethal in homozygous state. The main sign of the MDM manifestation and accumulation in Drosophila is mortality of organism from embryo to eclosion of imago from pupae[13]. In adult flies, MDMs affect individual adaptation to the environment. In such experiments, a reduction in viability and partial mortality of the progeny increases in 20[14], 40[15], 250[16] generations. These MDMs appear practically in each individual in the progeny at a rate an order of magnitude higher than that of recessive lethals and manifest in heterozygotes with the coefficient of dominance 0.2-0.5[15],[17]. Our approach was to study the progeny viability in the strains of Drosophila carrying recessive meiotic mutations (mei-mutations) maintained using balancer chromosomes or transmitted from father to son. In this case, selection for reproduction of only heterozygotes with balancers is not required for MDM accumulation. Mei-mutations in homozygote do not affect viability of their carriers. They only disturb meiosis, thus affecting fertility (due to the formation of abnormal gametes) and promote MDM preservation and accumulation in the progeny of small laboratory populations. The absence of recombination in male meiosis upon only paternal X-chromosome inheritance also promotes MDM accumulation.The purpose of this paper is to clarify the nature and meсhanism of MDM elimination at meiosis.
2. Materials and Methods
2.1. Fly strains
- Flies were reared at 25°C on standard medium. We used the following strains of Drosophila melanogaster (the abbreviated designation is given):(1) st[1] c(3)G17 [1] ca[1]/ TM2 ri Ubx[130] e[s] ca[1] (c(3)G17/TM2); (2) st[1] c(3)G17 [1] ca[1]/TM3 y+ri[1] p[p] sep bx[34e] e[s] Sb[sbd-1] Ser[1] (c(3)G17/TM3); (3) sp[2];st[1] c(3)G17 [1] ca[1]/ TM1 Me[1] kni[ri-1] Sb[sbd-1] (c(3)G17/TM1); (4) ru[1] h[1] th[1] st[1] cu[1] sr[1] e[s] ca[1]/ TM6 Hu[1] e[1] Tb[1] ca[1] ] (rucuca/TM6 Tb); (5) h[1] th[1] st[1] cu[1] sr[1] e[s] Pr[1] ca[1]/ TM6B Bri[1] Tb[1] (TM6B Bri Tb); (6) ru[1] h[1] th[1] st[1] cu[1] sr[1] e[s] ca[1]; (7) y[1]; al[1] dp[1] b[1] pr[1] cn[1] mei-W68[L1]/In(2LR) SM1 al[2] Cy cn[2] sp[2] (Cy suppressed) (mei-W68/SM1); (8) y[1] w[1]/Dp(1;Y) y[+]; mei-P22[P22]; sv[spa-pol] (mei-P22/); (9) Dp(1;1) sc[V1] y[1] mei-41[1] car[1] y[+]/C(1)DX y[1] f[1] bb[-]/Y (not y[+]) (mei-41/); (10) Dp(1;1} sc[VI] y[1] mei-218[1] car[1] y[+]/C(1)DX y[1] f[1] bb[-]/Y (mei-218/); (11) l[21pn]/FM4 y[31d] sc[8] dm[1] B[1] (/FM4); (12) Df(2L)A267, b cn bw/In(2LR)O Cy dp pr cn (/CyO); (13) wild type (Oregon R) (+/+).The information on the genome, mutations, and balancers of D. melanogaster is presented in the manual by Lindsley and Grell[18] and at http://flybase.bio.indiana.edu.
2.2. The Method of Examining the Viability of the Heterozygous Parent’S Progeny
- The crossing of heterozygous parents excludes the effects of recessive mei-mutations in the progeny (Fig. 1).
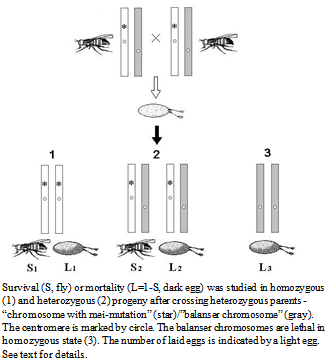 | Figure 1. Scheme of studying the effect of MDMs accumulation on the viability of the progeny |
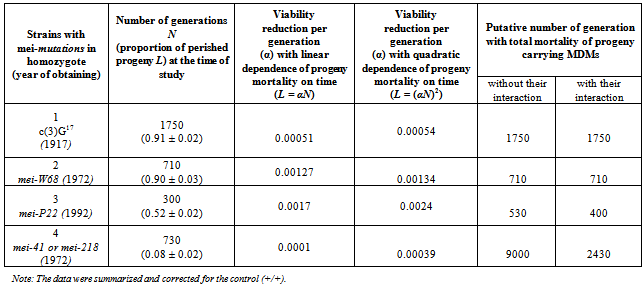
2.3. Counting the Number of Generations, Accumulating MDMs
- The number of generations (N), during which there were violations of the pairing and recombination of chromosomes during meiosis under the influence of different mutations, was calculated. Strains with mutations were maintained with crossover suppressors from the time of their registration or production to the time of the experiments. The strains carrying mei-с(3)G17, mei-W68, mei-P22, mei-41 and mei-218 are maintained since 1917[19],1972[20], 1992[21], 1972[22] respectively. Taking into account the duration of the Drosophila life cycle and the practice of maintaining strains in laboratory (18 days), consequently, the approximate number of generations elapsed to the time of analysis were different for different strains: 1750 for mei- с(3)G17, 710 for mei-W68, 300 for mei-P22, 730 for mei-41 and mei-218.
2.4. Viability of Progeny in Strains and Their Hybrids Carrying Mei-Mutation C3)G17
- Progeny viability was examined in three laboratory strains of D. melanogaster (1-3), carrying meiotic mutation c(3)G17. Laboratory strain 2 (c(3)G17/TM3) is maintained since 1985; it was derived from strain 1 (c(3)G17/TM2) by substitution of the balancer. Strain 2 was supplied by I.D. Alexandrov (United Institute of Nuclear Research, Dubna, Russia). Strain 3 (c(3)G17/TM1) was provided by the Bloomington Drosophila Stock Center, previously, it was kept in the Caltech Stock Center, approximately up to 1970 (http//www.flybase.edu). In addition, we examined viability of the hybrid progeny, homozygous for the mei-mutation and produced by crossing flies of different strains (1 × 2, 1 × 3 and 2 × 3). To facilitate phenotypic marking of the progeny, we transferred the studied chromosome (carrying mutation c(3)G17) to heterozygote with a new balancer chromosomes; the flies were taken in the experiment during three generations. In some cases the reproduction was conducted for about 20 and 50 generations. We used strains 4 and 5 as a source of new balancer chromosomes and strain 6 for generating crossover chromosomes (females st c(3)G17 ca/ru h th st cu sr e ca) containing different regions with accumulated MDMs ( th st cu c(3)G17 (?) ca, ru h st c(3)G17 (?) sr e ca). Strain 13 was used as control. Strains 4–6, 13 were also provided by the Bloomington Stock Center.
2.5. Comparative Analysis of Viability in Strains Carrying mei-mutations c(3)G17, mei-P22, mei-W68, mei-41, mei-21
- Progeny viability was examined in D. melanogaster strains 7-10, homozygous at various mei-mutations (mei-W68, mei-P22, mei-41, mei-218). Strains 7 and 9 were provided by A.T.C. Саrpenter. In the strain 8 with mutation mei-Р22, the balancer chromosome was at some point lost, and this mutation is currently maintained in homozygote (http://flybase.org).Strains 8 and 10 were supplied by the Bloomington Stock Center. Strains 4 and 11 were used as a source of new balancer chromosomes.
2.6. Mortality of Hybrid Progeny with Chromosomes From Strains Carrying Different Mei-Mutations with Accumulated MDMs
- Strains 7, 8, 2 -, containing the mei-mutations W68, P22 and c(3)G17 respectively were used. Strains 4 and 12 were used as a source of new balancer chromosomes. We examined the mortality of the following hybrid progeny: (a) c(3)G17/ mei-P22; (b) mei-W68/+, c(3)G17/+ and (c) mei-W68/+, mei-P22/+. In addition, mortality of the progeny was studied in heterozygotes c(3)G17/rucuca, c(3)G17/TM6Tb, mei-P22/TM6Tb and mei-W68/SM1.
3. Results and Discussion
3.1. Viability of the Progeny in Strains and Their Hybrids Carrying Mei-mutation c(3)G17
- In this section of paper we refer to some historical research data. Since the average value of life cycle duration estimated previously[13],[23] was somewhat lower, here it was standardized for comparing different strains.The first meiotic mutation, с(3)G17, was found in a natural population in 1917 and has been since then maintained in laboratory strains. Mei-mutation с(3)G17 in autosome 3 (3-57.4; 89A5) disturbs the formation of synaptonemal complex (SC), suppresses recombination in homozygous females[24],[25], and, which is worth mentioning, enhances recombination in heterozygotes with the normal chromosome[26]. Note also the exclusive maintenance of autosome 3 in all strains in heterozygote at balancers and the absence of chromosome pairing and crossing over in homozygotes due to the mei-mutation.
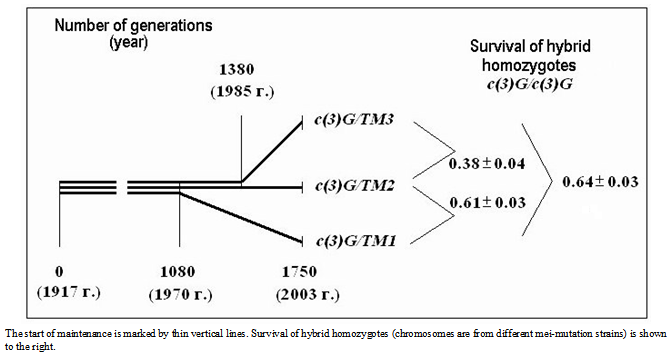 | Figure 2. History of strains carrying mei-mutation c(3)G17 |
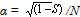 . Considering the lethality of the progeny in wild-type strain +/+ (0.09 ± 0.02),
. Considering the lethality of the progeny in wild-type strain +/+ (0.09 ± 0.02), 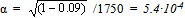 . The factual data correspond to the splitting of the strains from the initial stock in 1970 and 1985, i.e., after their maintenance together for 1010 and 1430 generations, respectively. The above relationship suggests that MDMs interaction corresponds to their synergistic epistasis.
. The factual data correspond to the splitting of the strains from the initial stock in 1970 and 1985, i.e., after their maintenance together for 1010 and 1430 generations, respectively. The above relationship suggests that MDMs interaction corresponds to their synergistic epistasis. 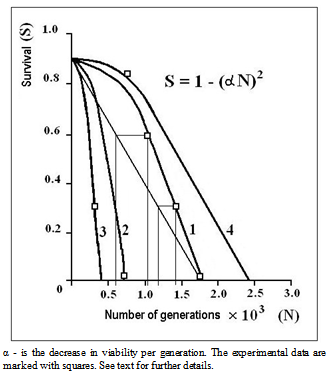 | Figure 3. Putative quadratic relationship between the progeny viability S and the number of generations N that accumulate MDMs in strains carrying mei-mutations c(3)G17 (1), W68 (2), P22 (3), 41 or 218 (4) |
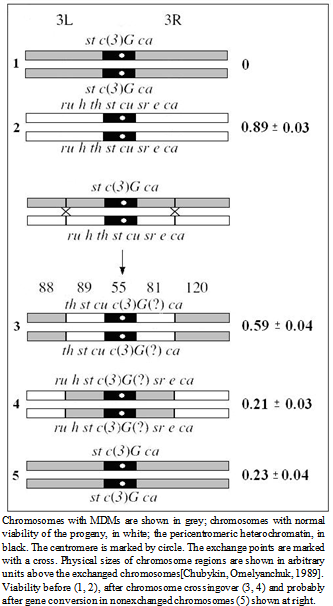 | Figure 4. Viability of homozygous progeny before and after mei-recombination in heterozygotes st c(3)G17 ca/ru h th st cu sr e ca |
3.2. Comparative Analysis of Viability of the Progeny in Strains Carrying Mei-mutations c(3)G17, mei-P22, mei-W68, mei-41, mei-218
- To evaluate the role of other meiotic events that promote MDM accumulation, we examined the accumulation of MDMs in strains having other mei-mutations: mei-W68 (2-94; 56D9) and mei-P22 (3-21.5; 65E9). Both these mutations disturb the formation of double-strand DNA breaks without changing SC[21],[33]. In addition, we studied strains carrying mei-mutations in the X chromosome (mei-218 (1-56.2, 15D6) and mei-41 (1-54.2; 14C3)), disrupting repair of DNA breaks appearing during meiosis[20]. We note that mutations at the time of their generation and description did not affect the viability of their carriers in either homozygote or heterozygote. The effect of mei-mutations on MDM accumulation in a laboratory strain is possible only in homozygotes for recessive mei-mutations. This condition was met in laboratory strains carrying mutations с(3)G17, mei-W68 and mei-P22.
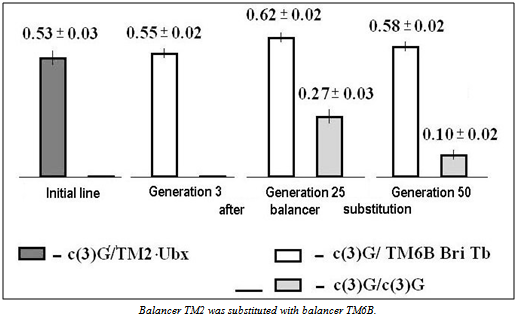 | Figure 5. Effect of balancer replacement on the viability of heterozygotes and homozygotes for mei-mutation c(3)G17 depending on the number of generations |
|
|
3.3. Mortality of the Hybrid Progeny With Chromosomes from the Strains Carrying different Mei-Mutations with Accumulated MDMs
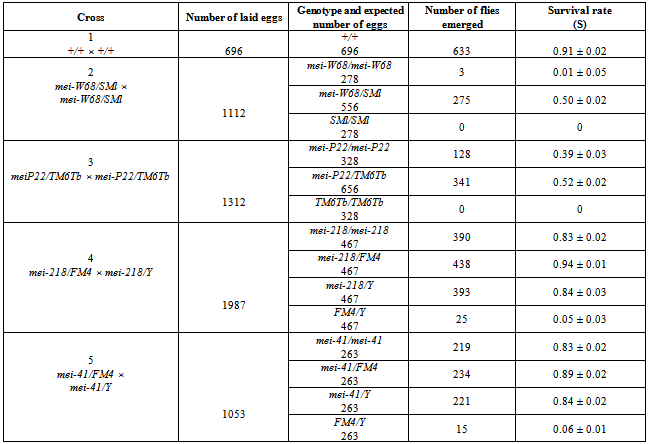
- The MDM effect on viability of hybrids containing heterozygous (from different strains) chromosomes with MDMs in autosome 3 (cis position) with c(3)G17/ mei-P22 was examined (Fig. 6a). We also examined hybrids with combination of autosomes 2 and 3 with MDMs from strains with meiotic mutations c(3)G17, mei-P22 and mei-W68 in heterozygote with a chromosome that conferred normal viability (trans position), mei-W68/+, c(3)G17/+ (Fig. 6b) and mei-P22/+, c(3)G17/+ (Fig. 6c). The results produced in different crosses are presented and analyzed in Fig. 6 and Table 3. The mortality of the progeny of the wild-type strain +/+ (0.09 ± 0.03) and homozygotes for multiple phenotypic markers rucuca/rucuca (0.11 ± 0.02) showed practically no difference (P<<0.95). The high mortality of progeny rucuca/TM6Tb (0.42 ± 0.03 and 0.47 ± 0.03 in different crosses) suggests that the balancer chromosomes, in addition to lethal mutations, carry MDMs]. However, the equal mortality of the heterozygous progeny with c(3)G17/rucuca and c(3)G17/TM6Tb (0.57 ± 0.03 and 0.57 ± 0.03 respectively, Table 3) cast doubt on this assumption. Nevertheless, these results indicate that there is no difference between the mortality of heterozygous progeny with chromosomes carrying accumulated MDMs in balancers and with structurally normal chromosomes, which was established earlier in classical studies[14],[15].
|
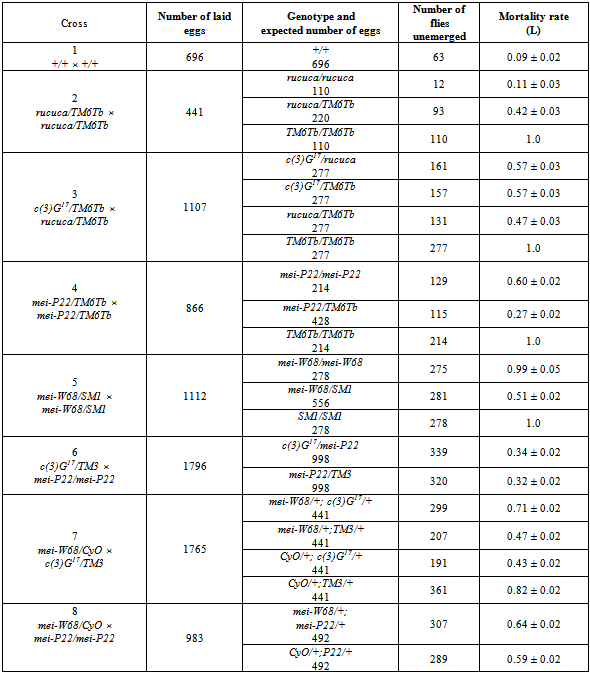
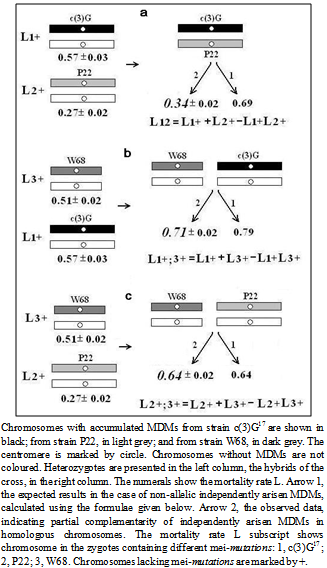 | Figure 6. Mortality rate of heterozygotes and hybrid progeny carrying homologous (a) and nonhomologous (b, c) chromosomes with independently accumulated MDMs in different mei-mutant strains |
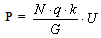 | (1). |
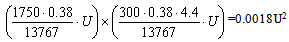 | (2) |
3.4. The Nature of MDM
- The nature of MDMs is still unclear. The MDM expression is similar to the position-effect variegation - all progeny carries MDMs but only part of the progeny perishes. This similarity in mutation expression also suggests that MDM impair the gene activity regulation. We have shown that normal meiosis in heterozygote of chromosomes with accumulated MDMs and structurally normal, MDM-free chromosomes restores viability both in recombinant and in non-crossover chromosomes with accumulated MDMs (Fig. 4). Chromosomal recombination as such apparently does not significantly reduce MDM pressure, since we cannot exclude the involvement of gene conversion (intragenic recombination), which is also initiated by DNA breaks [30-32]. This is evidenced by the data on partial viability restoration during 20 generations by changing the balancer (Fig. 5). The MDM elimination in meiosis is problematic to explain on the basis of gene nature of MDMs, changing DNA sequence, since the probability of their reverse mutation is very low. All facts listed above suggest that MDMs in some manner change gene expression rather than changing DNA.In contrast to the effect of mutation c(3)G17, the mei-mutations examined in this study (mei-W68, mei-P22) do not disturb homologous chromosome pairing and the SC formation. These mutations affect the initiation of recombination[21],[33] by means of impairing the generation of DNA breaks, produced by topoisomerases after the SC formation in Drosophila oocytes. It is known that DNA breaks, releasing structural tension in the packaged chromatin, promote its reorganization and accessibility in structural modification[34] (for instance, in the processes of inter- and intragenic recombination and DNA repair). Thus, the results of the present study do not contradict our suggestion of the epigenetic nature of MDMs, disturbing the formation and inheritance of specific functional structures of the genome[35]. These are probably structural-functional chromosome domains arranged in chromatin loops and containing gene clusters comparable with their putative number (≈ 17). The number of the loops is nearly by one order of magnitude lower than that of the genes (approximately 13767/17 ≈ 810, where the numerator is the total number of genes and the denominator, the calculated average number of genes per loop). According to the DNA content in female chromosomes[29], the numbers of loops in the X chromosome and autosomes 2, 3, and 4 are respectively 187, 286, 309, and 28.The chromatin loops are functional and structural chromosome units responsible for gene expression, replication and recombination in eukaryotes[36]. The loops are of different size. In the telomeric and pericentromeric chromosome regions, where the rates of recombination and MDM accumulation are highest (Fig.4), the loops are most abundant, but smaller than in other chromosome parts[37]. The repeated DNA sequences at the loop base are highly variable and associated with the chromosome axial element or nuclear matrix (S/MARs). Similar sequences were detected in loops beyond the axial element, where they may act as potential triggers for the formation of new domains [38],[39]. Apparently, their secondary structure (hairpins made of inverted repeated sequences) is essential for their functioning (anchoring on the axis and forming the loops) (Fig. 7)[40]. The loops size range from 3 to 200 kb; the size of interloop stretches also vary, but in the narrower range (from 3 to 30 kb)[41]. FISH visualization of DNA probes on preparation of nuclei extracted by 2M NaCl (nuclear halos) showed that active genes are localized in the loop bases harboring the complexes for replication, transcription, DNA repair and recombination, where various topological problems of the chromatin are resolved[42]. Most results obtained using different methods of DNA loop mapping, indicate their nonrandom standard organization [43]. The issue on association of the loop structure with epigenetic regulation of gene expression, including genetic imprinting and gene-position variegation, with meiotic chromatin remodeling has long been discussed[44],[45]. The loop domains linearly and spatially border a large group of genes, ensuring interaction enhancers and promoters within these limits, i.e., act also as insulators[46-48]. The major properties of loop domains are shown in (Fig. 7).
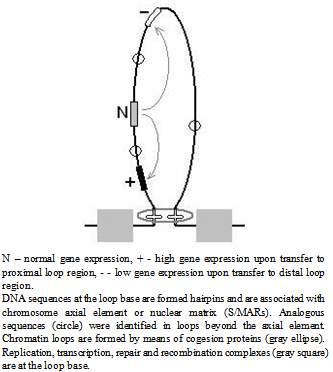 | Figure 7. Relationship between gene activity and gene position in the chromatin loops |
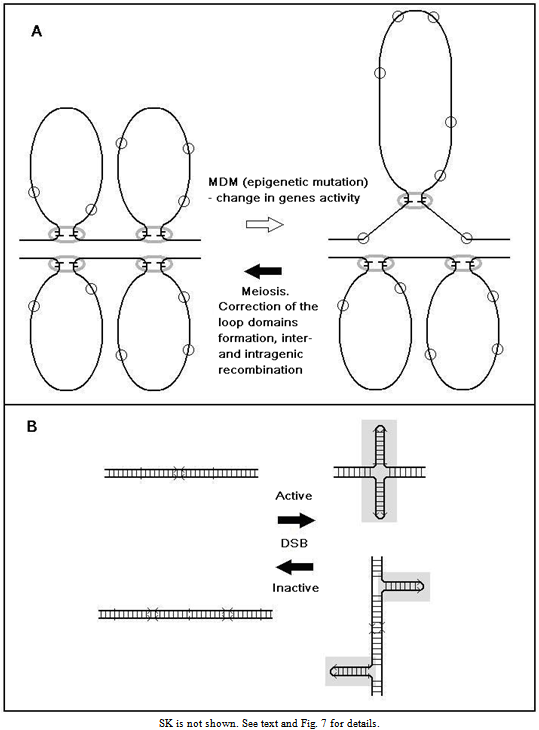 | Figure 8. The directional correction of the loop domain formation in meiosis (A) is accompanied by the rearrangement of the secondary structure of S/MAR repeats (B) by means of double-strand DNA breaks (DSB) |
4. Conclusions
- Thus, the relationship between survival and generation number upon MDMs accumulation suggests that MDMs interaction corresponds to their synergistic epistasis, that impair the formation of DNA breaks is more effective in accumulation of MDMs. The viability in progeny after meiosis in heterozygotes with chromosome with accumulated MDMs and normal chromosome, and in heterozygotes with independently accumulated MDMs chromosomes was shown to be partially restored (20% and 50% respectively). Our results support the hypothesis that MDMs have epigenetic nature. The potential polyvariant character of the chromatin loop formation and the existence of genes with the expression decreasing from the loop base, taken together with our results unambiguously support our conclusion on the nature of MDMs. The sexual reproduction is important for generating the evolutionary potential of the species and its further evolution, but it’s most vital function is conferring stability to the species by means of meiosis. In essence, resolving this issue will provide insight in understanding evolution, which implies increasing the complexity of the structural organization of life upon the presence of nearly equal standard gene set in the majority of higher organisms. The simple argument for epigenetic nature MDM may be obtained through using the method of comparative DNA loop by mapping a number of genes in wild chromosomes and chromosomes carrying mei-mutations.
ACKNOWLEDGEMENTS
- The author is very grateful to Ivan P. Glushkov, T.M. Grishaeva and A.G. Imasheva for technical assistance. The study was supported by the Russian Foundation for Basic Research (grant nos. 05-04-49052, 08-04-01725a).