-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Stem Cell Research
2012; 1(1): 1-8
doi: 10.5923/j.ajscr.20120101.01
Differential Release of Heterogeneous Human Mesenchymal Stem Cell Populations from Haemarthrotic Traumatic Knee Injury
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLDavid J. Deehan 1, 2, Daniel J. Dowen 2, Andrew P. Sprowson 1, Linda C. Ferguson 1, Nilendran S. Prathalingam 3, John D. Isaacs 1, 2, Mark A. Birch 1, Rachel A. Oldershaw 1
1Institute of Cellular Medicine, Faculty of Medical Sciences, Newcastle University, Medical School, Framlington Place, Newcastle-upon-Tyne, NE2 4HH, UK
2Freeman Hospital, Newcastle-Upon-Tyne NHS Trust, Freeman Road, High Heaton, Newcastle-upon-Tyne, NE7 7DN, UK
3Institute of Ageing and Health, Faculty of Medical Sciences, Newcastle University, International Centre for Life, Times Square, Newcastle-upon-Tyne, NE1 4EP, UK
Correspondence to: Rachel A. Oldershaw , Institute of Cellular Medicine, Faculty of Medical Sciences, Newcastle University, Medical School, Framlington Place, Newcastle-upon-Tyne, NE2 4HH, UK.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Surgical reconstruction of the anterior cruciate ligament (ACL) has a protracted healing phase due to poor osseous tissue integration at the graft/host interface. Intervention with an autologous cell-based therapy using human mesenchymal stem cells (hMSCs) derived from haemarthrotic fluid aspirated at the acute phase of injury has been postulated to accelerate healing, though until now the practicalities of this approach have not been demonstrated. hMSCs were derived by plastic adherence from haemarthrosis fluid aspirated from 20 patients presenting at clinic with acute knee injury. Patient details were recorded including age and sex of patient, injury and time between injury and aspiration. The phenotype of hMSCs was characterised by flow cytometry analysis of cell surface antigens. Differentiation potential was analysed by culturing hMSCs with different pro-differentiation stimuli to drive osteogenesis, adipogenesis and chondrogenesis. Comparative analysis of differentiation was made by quantitative PCR for lineage-specific gene expression and quantitative biochemical analyses. hMSC derivation was independent of age, sex and time between injury and aspiration however there was a statistically significant increase in frequency of derivation from haemarthosis samples that had been aspirated from bone fracture injuries compared to soft tissue injuries. hMSCs showed differential expression of cell surface antigens and there were also significant differences in their osteogenic, adipogenic and chondrogenic responses between samples. We have demonstrated the feasibility of deriving multipotent hMSCs from haemarthrosis fluid aspirated from acute knee injuries. Further optimisation of processing and differentiation methodologies must be achieved to develop a feasible clinical treatment which accelerates ACL reconstruction. This study has identified challenges in the harvesting, bio-processing and characterisation of hMSCs which would be broadly applicable to the development of all autologous orthopaedic cell therapies.
Keywords: Haemarthrosis, Mesenchymal Stem Cell, Multipotent Differentiation, Acute Knee Trauma
Article Outline
1. Introduction
- Injury and rupture of the anterior cruciate ligament (ACL) as a result of trauma is common and results in acute bleeding into the knee joint[1]. The reconstruction of the ligament requires the tethering of an autograft inside a bony tunnel and the successful surgical outcome of this technique is wholly dependent upon tissue integration and remodelling to enable strong anchorage at the graft/host interface[2]orthopaedic tissues including cartilage, bone, tendon and meniscus[3-6]. Within the context of ACL reconstruction hMSCs potentially offer a new autologous therapeutic modality to accelerate tissue repair, graft integration and restoration of normal knee function. Whilst commonly being derived from the stromal compartment of bone marrow hMSCs have been sourced from diverse tissues including adipose, trabecular bone, cartilage, skeletal muscle, peripheral blood and synovial fluid[7-10]. Analgesic aspiration of an acute knee effusion could therefore allow for harvesting of blood and tissue fluid with a view to isolating hMSCs[11, 12]. This presents a practicable clinical model by which hMSCs are derived, expanded and assembled into an autologous deliverable product during the 10-12 week period required for stabilisation of the knee joint prior to reconstructive surgery. We wished to examine the feasibility of reproducibly harvesting and isolating hMSCs from haemarthroses and demonstrate their multipotential with the longer term view of their introduction with a scaffold to enhance tissue repair. We have investigated the derivation of hMSCs from a consecutive series of 20 patients who presented at clinic with a diverse range of acute knee trauma. hMSC populations were characterised for expression of key cell surface antigens and the potential to undergo in vitro differentiation to chondrocytes, osteoblasts and adipocytes. We report our experience and outline the early difficulties encountered in advancing this therapeutic approach.
2. Materials and Methods
2.1. Acquisition of haemarthrosis fluid
- All patients gave informed consent according to Ethical Committee guidelines. A consecutive series of patients presenting with a traumatic knee haemarthrosis underwent aspiration under aseptic conditions. Samples were collected in heparinised specimen tubes and immediately stored at 4ºC. Samples were processed within 24 hours of aspiration. Clinical diagnoses as suspected through clinical examination were confirmed by plain radiography and MRI scanning.
2.2. Cell Culture
- All cell culture reagents were purchased from Invitrogen, Paisley, UK unless otherwise stated.
2.2.1. Derivation and culture of hMSC
- Hemarthrosis fluid was diluted with DPBS at a ratio of 1:4, layered onto a 12ml volume of Ficoll-Paque™ Premium (GE Healthcare Life Sciences, Buckinghamshire, UK) and centrifuged at 600 x g for 20 minutes. The mononuclear cell fraction was centrifuged at 25,000 x g for 5 minutes and the cell pellet resuspended in hMSC medium (Minimum Essential Medium (MEM) α medium, 10%(vol/vol) FBS, 2mM GlutaMAX™, 5ng/ml FGF2) and seeded in a T-75 cell culture flask at a density of 300 x 105 cells / cm2. After 24 hours culture medium was removed and the culture area rinsed with DPBS. Bone marrow mononuclear cells (Lonza, Wokingham, UK) were seeded in a T-75 cell culture flask at a density of 300 x 105 cells / cm2 and cultured in hMSC medium as previously described[13].
2.2.2. Osteogenic differentiation
- hMSCs were seeded cultured with osteogenic medium (Dulbecco’s Modified Eagles Medium (DMEM), 10% (vol/vol) FBS, 2mM GlutaMAX™, 10mM β-glycerophosphate, 10nM dexamethasone, 100nM L-ascorbic acid-2-phosphate). Cultures were maintained for 4 weeks[14].
2.2.3. Chondrogenic differentiation
- hMSC were detached from monolayer and resuspended in chondrogenic medium(Dulbecco’s Modified Eagles Medium (DMEM), 2mM GlutaMAX™, 100nM dexamethasone, 100nM L-ascorbic acid-2-phosphate, 40µg/ml L-proline, 1% (vol/vol) ITS+1, 10ng/ml TGF-β3). Cell aggregates (5 x 105 cells / aggregate) were formed by centrifuging the tubes at 300 x g for 5 minutes. Cultures were maintained for 2 weeks[13].
2.2.4. Adipogenic Differentiation
- Adipogeneic differentiation was initiated by cyclical treatment of 72 hours culture with adipogenic induction medium (Dulbecco’s Modified Eagles Medium (DMEM), 10% (vol/vol) FBS, 2mM GlutaMAX™, 1% (vol/vol) ITS+1, 1µM dexamethasone, 100µM indomethacin, 500µM 3-isobuty-1-methylxanthine) followed by 24 hours culture with adipogenic maintenance medium (Dulbecco’s Modified Eagles Medium (DMEM), 10% (vol/vol) FBS, 2mM GlutaMAX™, 1% (vol/vol) ITS+1) . After 4 cycles cells were cultured for 7 days in adipogenic maintenance medium[14].
2.3. Flow Cytometry
- hMSC populations was analysed by flow cytometry[15]; primary antibodies(5µg/ml) were taken from the Human Multipotent Mesenchymal Stromal Cell Marker Antibody Panel (R&D Systems, Minneapolis, USA, secondary antibody (donkey anti-mouse IgG Alexa 488; 8µg/ml (Molecular Probes, Invitrogen)). Cell labelling was analysed with a BD FACSCanto™ II flow cytometer and BD FACSDiva™ software (BD, Oxford, UK).
2.4. Gene Expression Analysis
- Total RNA was prepared from monolayer cell cultures using Tri-reagent. Total RNA was prepared from chondrogenic cell aggregates using Tri-reagent in conjunction with the Molecular Grinding Resin™ with Pestle and Mortar kit (GBiosciences, St Louis,USA). Total RNA(1µg per reaction) was reverse transcribed using 500ng random hexamers, 500µM dNTPs, 200U M-MLV reverse transcriptase and 1 X reverse transcription reaction buffer (Promega, Southampton, UK). Quantitative PCR(qPCR) was performed with the qPCR™ Core Kit for Sybr™ Green I. Each qPCR reaction was assembled to a total volume of 25µl using 1µl cDNA template, 300nM gene-specific forward and reverse primer set, 1 X PCR reaction buffer, 3.5mM MgCl2, 200µM dNTPs, 0.625U HotGoldStar enzyme, 1:66000 Sybr Green I. Gene-specific forward and reverse primer sets were as previously published; GAPDH[16], AP2 and LIPOPROTEIN LIPASE[17], CBFA1 and ALKALINE PHOSPHATASE[18]. Data was normalised to GAPDH using the delta-delta method[19].
2.5. Histological Analysis
2.5.1. Safranin O stain of chondrogenic cell aggregates
- Chondrogenic cell aggregates were fixed in 4% (vol/vol) formaldehyde, embedded in paraffin wax and sectioned (5µm). Deparaffinised sections were stained in Harris’ haematoxylin, stained with 0.02% (wt/vol) aqueous fast green FCF for 3 minutes, and stained with 0.1% (wt/vol) aqueous safranin O for 5 minutes. Slides were mounted using DePeX mounting medium.
2.5.2. Alizarin red stain of osteogenic cultures
- Osteogenic cultures were stained with Alizarin Red (2% wt/vol, pH4.2) for 2 hours at room temperature. Quantification of alizarin red staining was carried out by solubilising cell cultures with 2% (wt/vol) SDS and measuring the absorbance (A492) using a Multiskan Ascent plate reader (MTX Lab Systems, Inc., Vienna, USA).
2.6. Biochemical Analysis
2.6.1. Preparation of Cell Lysates
- Monolayer cell cultures were lysed in 2ml volumes of 0.01% Triton-X-100. Chondrogenic cell aggregates were digested in 20µl of 10U/ml of papain in papain buffer (100mM sodium acetate, 2.4mM EDTA, 5mM L-cysteine (pH5.8)) at 60°C.
2.6.2. Quantification of DNA
- Total DNA in the prepared cell lysates was measured using the Quant-it™ PicoGreen® dsDNA Assay kit. Fluorescence was measured using a Fluoroskan I plate reader (MTX Lab Systems, Inc.).
2.6.3. Metachromatic Quantification of Sulphated Glycosaminoglycans
- Quantification of sGAG was carried out by 1,9-dimethylmethylene blue (DMMB) assay. Experimental samples were mixed with DMMB solution (0.16% (wt/vol) DMMB, 0.2% (vol/vol) formic acid, 30mM sodium formate (pH 3.5)) and the absorbance(A595) read using a Multiskan Ascent plate reader(MTX Lab Systems, Inc.). Values were calibrated against known concentrations of shark chondroitin sulphate.
2.7. Statistical Analysis
- Two-sample Student’s T-tests were carried out on data sets that were seen to be normally distributed. Where data was not normally distributed non-parametric equivalents were used.
3. Results
3.1. Derivation of hMSC Populations from Haemarthrosis Fluid
- We compared the ability to derive hMSC populations from haemarthrosis fluid that had been aspirated from 20 consecutive patients presenting at clinic with a diverse rangeof acute knee trauma(Table 1). The study group comprised11 male and 9 female patients aged between 15-79 years with the average age being 48 years and the median age 52 years. Eight of the twenty acute knee injuries were resultant of soft tissue ruptures where bleeding into the synovial joint space would occur solely from the peripheral circulation. Bone fractures accounted for twelve of the twenty acute knee injuries and as such the aspirated haemarthosis would contain a further molecular and cellular contribution from the bone marrow compartment of the osseous tissue. For the majority of samples aspiration of the haemarthrosis took place within 1 day of injury. Five of the samples were aspirated within 1 week and the remaining around 2 weeks (sample 3, 15 days; sample 14, 14 days) post-injury.We observed a significant amount of haemolysis within some of the samples such that we were frequently unable to resolve clear phase separation of the mononuclear cell fractions following ficoll-gradient centrifugation. Colony-forming unit fibroblasts(CFU-Fs) with characteristic hMSC morphology were observed between 7-14 days post-seeding from twelve out of twenty samples. The number of CFU-Fs obtained from each sample was independent of the amount of fluid that was aspirated. The ability to derive hMSC populations was independent of gender, age and time between injury and aspiration; however there was a significant difference between rate of derivation and the origin of haemarthrosis. We derived hMSC populations from 9 out of the 12 samples that had been aspirated following bone fracture injuries compared to three of the eight haemarthroses arising from soft tissue ruptures (P<0.005).
3.2. Human Mesenchymal Stem Cell Populations Derived from Aspirated Haemarthroses Display Heterogeneric Cell Surface Antigen Expression between Samples
- Populations of hMSCs that had been successfully derived from aspirated haemarthroses were expanded to passage 2 in in vitro culture and analysed by flow cytometry for the expression of characteristic hMSC cell surface antigens (Figure 1). Data is omitted for samples 2, 8 and 12 where we were unable to generate sufficient cell numbers to carry out flow cytometry analysis alongside multipotent differentiation assays. The proportion of hMSCs expressing each of the cell surface antigens was highly heterogeneous between samples. In general, a high proportion of each cell population was seen to express CD44, CD90, CD105 and CD166. CD44 was expressed on over 94% of hMSC populations with the exception of samples 3, 1 and 17. CD90 was expressed on over 95% of hMSC populations with the exception of samples 6, 3 and 18. CD105 was expressed on over 95% of hMSC population with the exception of samples 11, 3, 17 and 6. The cell surface antigen CD166 was expressed on a relatively high proportion of cells(>80%) within hMSC populations derived from samples 1, 11, 17, 18 and 20. The proportion of cells expressing the cell surface antigens
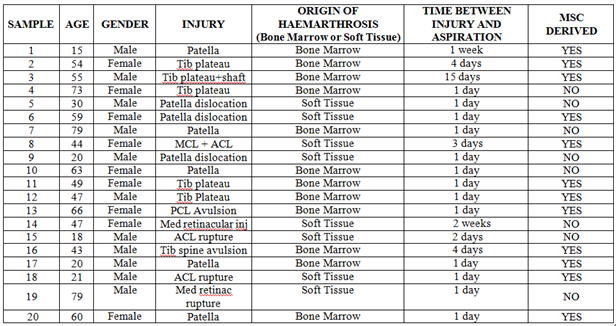
 | Figure 1. Flow cytometry analysis of hMSC populations. Human mesenchymal stem cell cultures that had been derived and expanded to passage 2 were analysed for expression of a panel of cell surface antigens characterised to be either present or absent on hMSCs. High numbers of cells expressing positive markers confirmed that we had derived hMSCs however the relative abundance of individual cell surface antigens varied considerably revealing highly heterogenic expression between samples. The detection of CD19 and CD45 expressing cells within some cell populations showed contamination with non-hMSC cell types |
3.3. Human Mesenchymal Stem Cell Populations Derived from Haemarthroses Exhibit Variation in Multipotent Differentiation between Samples
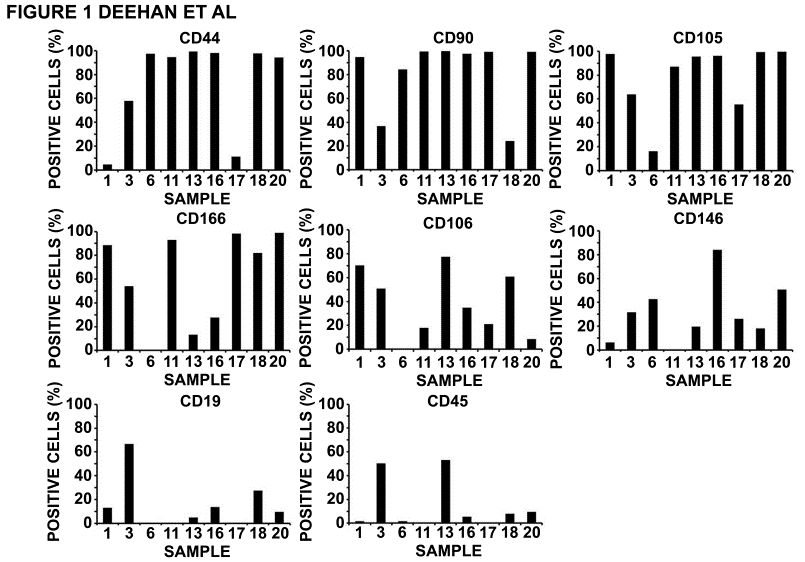
- To investigate the potential for multipotent differentiation, hMSC populations that had been successfully derived from haemarthrosis samples were expanded to passage 2 before being cultured in vitro with appropriate stimuli to promote differentiation to osteoblast, adipocyte and chondrocyte cell lineages[11,12]. All twelve of the hMSC populations that had been derived from haemarthrosis samples underwent osteogenic differentiation. At day 0 hMSCs that had been seeded at low cell density were seen to have a characteristic fibroblastic morphology. After 28 days of osteogenic differentiation significant cellular proliferation was evident. Cells had acquired an osteoblast-like cuboidal cell morphology and were encased within an extensive extracellular matrix (data not shown). CBFA1 and ALKALINE PHOSPHATASE, key markers of osteoblast phenotype, were expressed within all osteogenic cultures after 28 days though expression varied between hMSC populations by 100-fold and 1000-fold respectively for each gene(Figure 2a, b). Quantification of the amount of DNA was carried out at day 0 and day 28 as a measure of the amount of cellular proliferation that occurred during osteogenic differentiation(Figure 2c). For the majority of hMSC populations there was between 4 and 6-fold increase in DNA during osteogenic differentiation, indicative of cells within the culture having undergone 2-3 rounds of cell division. Some cell populations showed greater proliferative potential with 8 and 12-fold increases in the amount DNA(3-4 rounds of cell division) and 20 to 30-fold increases(5-6 rounds of cell division).Histological characterisation for calcified matrix was carried out by staining cell cultures with alizarin red (Figure 2d). Unsurprisingly at day 0 alizarin red staining was negative. In contrast, at day 28 there was significant alizarin red staining across the culture dish and this was apparent for all haemarthrosis-derived hMSC cultures that had between treated with pro-osteogenic stimuli. To provide a quantitative measure of matrix calcification, day 28 cultures that had been stained with alizarin red were solubilised and the absorbance read at A492. We have presented this as absorbance per amount of DNA within each sample; reflecting the contribution to matrix calcification per cell (Figure 2e). For the majority of hMSC populations the absorbance per DNA was between 0.07 and 0.1 units. However, hMSC populations derived from haemarthrosis samples 6 and 16 and those derived from samples 1, 2 and 18 showed approximately 3 and 4 times the amount of calcified matrix production per cell respectively.
 | Figure 2. Characterisation of osteogenic differentiation potential of hMSC populations. Human mesenchymal stem cell cultures that had been derived and expanded to passage 2 were analysed for their potential to differentiate to osteogenic, adipogenic and chondrogenic cell lineages. Osteogenic differentiation was assayed by gene expression analysis for (a) CBFA1 and (b) ALKALINE PHOSPHATASE (ALP) after 28 days in pro-osteogenic culture. (c) Cellular proliferation during osteogenic differentiation was recorded as the fold-change in the amount of DNA between cultures at day 0 and day 28. (d) Matrix calcification of osteogenic cultures was visualised by alizarin red staining of day 0 and day 28 cultures. (e) As a quantitative measure of matrix calcification, day 28 cultures that had been stained with alizarin red were solubilised and the absorbance read at A492. Alizarin red was recorded as a function of the total amount of DNA within the culture to indicate the contribution of calcification per cell. |
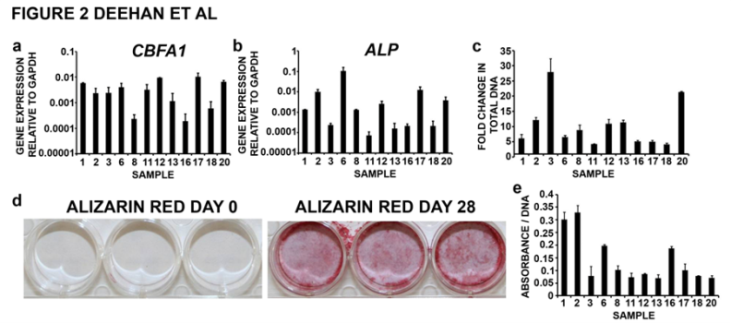
 | Figure 3. Characterisation of adipogenic differentiation potential of hMSC populations. Adipogenic differentiation was assayed by gene expression analysis for (a) AP2 and (b) LIPOPROTEIN LIPASE (LPL) after 30 days in pro-adipogenic culture. (c) Morphological analysis of cultures at day 30 of adipogenic differentiation showed enlarged cells with numerous mature lipid vesicles distributed though the cytoplasm; scale bar = 50µm |
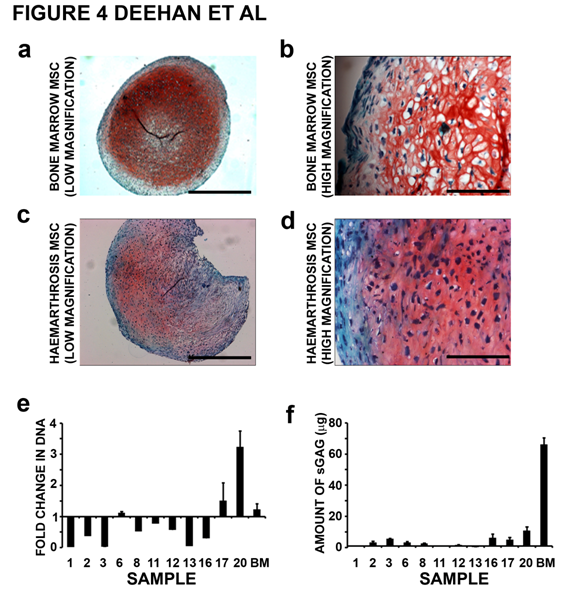 | Figure 4. Charcaterisation of chondrogenic differentiation potential of hMSC populations. Chondrogenic differentiation was analysed by safranin O staining of cells aggregate sections. (a) Images at low magnification revealed strong safranin O staining of cell aggregates formed from hMSCs derived from bone marrow; scale bar = 500mm. (b) At higher magnification rounded chondrocyte–like cells were shown to be encased within an extensive ECM; scale bar = 100µm. (c) Safranin O staining of cell aggregates formed from hMSCs derived from haemarthrosis fluid. Staining was weaker in comparison to that observed in cell aggregates formed from hMSCs derived from bone marrow and was also more heterogenic staining within the cell aggregates; scale bar = 500µm. (d) At high magnification rounded chondrocyte–like cells were shown to be encased within an extensive ECM; scale bar = 100µm. (e) Cellular proliferation during chondrogenic differentiation was recorded as the fold-change in the amount of DNA between cell aggregates cultures at day 0 and day 14. (f) Fold-change in the amount of sGAG accumulated between day 0 and day 14 of chondrogenic culture was measured by DMMB assay. BM = bone marrow. Values represent mean values ± S.E.M; n=3. |
4. Discussion
- The presence of hMSC populations within synovial fluid is well documented and their elevated numbers in response to disease and trauma demonstrates a role in the repair of damaged tissues within the synovial joint[8, 9, 20-22]. Our aim in this study was to investigate the potential of these cells as an autologous cell-based therapeutic modality. It has been argued that such cells may play a role in accelerating graft integration at time of surgical reconstruction[20-23]. This study accurately defines the potential yield, identifies the limitations in such work and begins to characterise the behaviour of such cells in relation to clinical characteristics. Only until such work is complete can this new technology become clinically and commercially deliverable.In this study hMSC populations were derived from 60% of haemathrosis aspirates that were collected from clinic. Derivation of hMSCs was independent of gender, time from injury to aspiration or the nature of the intra-articular event. Previous studies have found that hMSC derivation and culture is dependent on age[24, 25]. However, although we were unable to derive cells from the three most elderly patients, we were able to successfully obtain MSC populations from haemarthrosis fluid which had been aspirated from patients up to 66 years of age. The greatest influence on the ability to derive hMSCs was the origin of the haemarthrosis. We were significantly more successful at deriving hMSC populations from haemarthroses arising from bone fractures where there would have been an additional cellular component from the bone marrow compartment. From our observations and handling of the haemarthrosis samples, we would consider that the quality of the aspirate also influenced the ability to derive hMSCs. Despite patients being administered with heparin upon presentation at the clinic and also collection of the aspirate into heparin containing collection tubes there was a significant frequency of blood clotting and haemolysis between the aspiration and processing of the sample. This haemolysis appeared to compromise the ability to obtain clear phase separation from Ficoll-gradient centrifugation and resulted in contamination of the mononuclear cell population. We conjecture that this may have been a contributing factor in our inability to derive hMSCs from some heamarthrosis samples and will preclude the derivation of adequate cells for the generation of clinically deliverable product. Further understanding of the bio-processing procedures involved, such as aspiration technique, initial tissue handling and optimisation of the conditions used to store and transport the haemarthrosis fluid should reduce the frequency of haemolysis. We specifically characterised hMSC populations at passage 2 since we consider this to be the most appropriate point at which the cells would be used clinically. Using hMSCs at low passage should reduce the risk of deleterious effects brought about by adaptation to in vitro culture[26] whilst permitting the amplification of significant cell numbers for generating the clinical deliverable and use in mandatory testing as required by the appropriate regulatory agencies (Eudralex, Volume 4, Annex 2). Flow cytometry was carried out using a panel of commercially available antibodies specific for cell surface antigens expressed on hMSCs[6]. Differential expression of each antigen demonstrated that we derived highly heterogeneous populations; it is likely that these would become more homogeneous with prolonged culture[27, 28]. Whilst most antigens are expressed within high proportions of each cell population (CD44, CD90, CD105 and CD166) we noted much lower and more heterogeneous expression between cell populations when analysing expression of CD106 and CD146. Flow cytometry analysis will be one method by which we would carry out the necessary validation of cultured hMSC populations prior to clinical application. This data suggests that we will need to undertake a more considered approach in identifying a panel of appropriate antigens that will give us reliable characterisation of the hMSC phenotype with the minimum number of cells. In some samples there were large numbers of cells expressing the non-mesenchymal antigens, CD19 and CD45. It is likely that these cells would be lost during continued culture expansion though having argued strongly for the clinical application of these cells at low passage their exclusion from the final cell therapy must be addressed. We believe reduction of haemolysis will enable the isolation of cleaner mononuclear cell fractions and reduce lymphocyte contamination. It may be possible to introduce a negative selection technique to remove contaminating cells prior to incorporating the hMSCs onto the selected scaffold[29].Gene expression and biochemical analyses showed that we derived multipotent hMSC cells though there was a varied response to differentiation stimuli between cell populations. All of the twelve hMSC populations derived underwent adipogenic and osteogenic differentiation though some populations had a more robust proliferative and/or differentiation response than others. It is possible that for some samples(for instance sample 3) in which there were contaminating CD19+, CD45+ lymphocyte populations the cells had a more pro-inflammatory phenotype resulting in more proliferation over differentiation. Whilst there was evidence of chondrogenic differentiation within cell aggregates only one of the twelve hMSC populations showed true potential to differentiate along this lineage. Previous evidence has shown that the potential for differentiation toward cell lineages is restricted by tissue from which hMSCs are derived[30, 31]. For our intended aim of improved integration of the graft/host interface following ACL reconstruction, poor chondrogenic and adipogenic differentiation of our isolated hMSCs is not a concern. However, a more standardised response to osteogenic stimuli between different hMSC populations will need to be optimised. We predict improved global differentiation when the amount of non-hMSC cells has been reduced from starting cultures though the greatest benefit will be achieved through modification of pro-osteogenic culture conditions with particular emphasis on the microenvironment presented by the scaffold.
5. Conclusions
- We have demonstrated the feasibility of deriving hMSC populations from haemarthrotic fluid that has been aspirated in the acute clinical setting. Whilst clinical and scientific challenges remain we believe that these cells could fulfil a potential role in modulating the healing response to acute intra-articular injury.
ACKNOWLEDGEMENTS
- This work was funded by The JGW Patterson Foundation and from a NESCI Fellowship awarded to RAO. This work was supported by the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust. We wish to also acknowledge technical support provided by the flow cytometry facility, Newcastle University.
References
| [1] | Boden BP, Sheehan FT, Torg JS, Hewett TE. Noncontact anterior cruciate ligament injuries: mechanisms and risk factors. J Am Acad Orthop Surg 2010; 18: 520-7 |
| [2] | Deehan DJ, Cawston TE. The biology of integration of the anterior cruciate ligament. J Bone Joint Surg Br 2005; 87: 889-95 |
| [3] | Adesida AB, Grady LM, Khan WS, Hardingham TE. The matrix-forming phenotype of cultured human meniscus cells is enhanced after culture with fibroblast growth factor 2 and is further stimulated by hypoxia. Arthritis Res Ther 2006; 8: R61 |
| [4] | Hanson SE, Gutowski KA, P. Clinical applications of mesenchymal stem cells in soft tissue augmentation. Aesthet Surg J 2010; 30: 838-842 |
| [5] | Khan WS, Adesida AB, Tew SR, Lowe ET, Hardingham TE. Bone marrow-derived mesenchymal stem cells express the pericyte marker 3G5 in culture and show enhanced chondrogenesis in hypoxic conditions. J Orthop Res 2010; 28: 834-40 |
| [6] | Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284:143-147 |
| [7] | Jackson WM, Lozito T, Djouad F, Kuhn NZ, Nesti LJ, Tuan RS. Differentiation and regeneration potential of mesenchymal progenitor cells derived from traumatized muscle tissue. J Cell Mol Med 2010; E-pub ahead of print |
| [8] | Jones EA, Crawford A, English A, Henshaw K, Mundy J, Corscadden D et al. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: detection and functional evaluation at the single-cell level. Arthritis Rheum 2008 58: 1731-40 |
| [9] | Pei M, He F, Vunjak-Novakovic G. Synovium-derived stem cell-based chondrogenesis. Differentiation 2008; 76: 1044-56 |
| [10] | Sakaguchi Y, Sekiya I, Yagishita K, Muneta T. Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum 2005; 52:2521-259 |
| [11] | Lee SY, Miwa M, Sakai Y, Kuroda R, Matsumoto T, Iwakura T, et al. In vitro multipotentiality and characterization of human unfractured traumatic hemarthrosis-derived progenitor cells: A potential cell source for tissue repair. J Cell Physiol 2007; 210: 561-6 |
| [12] | Lee SY, Miwa M, Sakai Y, Kuroda R, Oe K, Niikura T, et al. Isolation and characterization of connective tissue progenitor cells derived from human fracture-induced hemarthrosis in vitro. J Orthop Res 2008; 26:190-9 |
| [13] | Oldershaw RA, Tew SR, Russell AM, Meade, Hawkins R, McKay TR, et al. Notch signaling through Jagged-1 is necessary to initiate chondrogenesis in human bone marrow stromal cells but must be switched off to complete chondrogenesis. Stem Cells 2008; 26: 666-74 |
| [14] | Barbero A, Ploegert S, Heberer M, Martin I. Plasticity of clonal populations of dedifferentiated adult human articular chondrocytes. Arthritis Rheum 2003; 48: 1315-25 |
| [15] | Oldershaw RA, Baxter MA, Lowe ET, Bates N, Grady LM, Soncin F, et al. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat Biotechnol 2010; 28: 1187-94 |
| [16] | Martin I, Jakob M, Schafer D, Dick W, Spagnoli G, Heberer M. Quantitative analysis of gene expression in human articular cartilage from normal and osteoarthritic joints. Osteoarthritis Cartilage 2001; 9: 112-8 |
| [17] | [17]Fink T, Abildtrup L, Fogd K, Abdallah BM, Kassem M, Ebbesen P, et al. Induction of adipocyte-like phenotype in human mesenchymal stem cells by hypoxia. Stem Cells 2004; 22: 1346-55 |
| [18] | Frank O, Heim M, Jakob M, Barbero A, Schafer D, Bendik I, Real-time quantitative RT-PCR analysis of human bone marrow stromal cells during osteogenic differentiation in vitro. J Cell Biochem 2002; 85: 737-46 |
| [19] | Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8 |
| [20] | Morito T, Muneta T, Hara K, Ju YJ, Mochizuki T, Makino H, et al. Synovial fluid-derived mesenchymal stem cells increase after intra-articular ligament injury in humans. Rheumatology (Oxford) 2008; 47: 1137-43 |
| [21] | Jones EA, English A, Henshaw K, Kinsey SE, Markham AF, Emery P, et al. Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis, Arthritis Rheum 2004; 50: 817-27 |
| [22] | McGonagle D, Jones E. A potential role for synovial fluid mesenchymal stem cells in ligament regeneration. Rheumatology (Oxford) 2008; 47:1114-16 |
| [23] | Ju YJ, Muneta T, Yoshimura H, Koga H, Sekiya I. Synovial mesenchymal stem cells accelerate early remodeling of tendon-bone healing. Cell Tissue Res 2008; 332: 469-78 |
| [24] | Coipeau P, Rosset P, Langonne A, Gaillard J, Delorme B, Rico A, et al. Impaired differentiation potential of human trabecular bone mesenchymal stromal cells from elderly patients. Cytotherapy 11:584-94 |
| [25] | Shamsul BS, Aminuddin BS, Ng MH, Ruszymah BH. Age and gender effect on the growth of bone marrow stromal cells in vitro. Med J Malaysia 59 Suppl B 2004:196-7 |
| [26] | Izadpanah R, Kaushal D, Kriedt C, Tsien F, Patel B, Dufour ,. et al. Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res 2008; 68: 4229-38 |
| [27] | Digirolamo CM, StokesD, Colter D, Phinney DG, Class R, Prockop DJ. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol 1999;107: 275-81 |
| [28] | Solchaga LA, Penick K, Porter JD, Goldberg VM, Caplan AI, et al. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol 2005; 203: 398-409 |
| [29] | Jarocha D, Lukasiewicz E, Majka M. Adventage of mesenchymal stem cells (MSC) expansion directly from purified bone marrow CD105+ and CD271+ cells. Folia Histochem Cytobiol 2008; 46: 307-14 |
| [30] | Kern S, Eichler H, Stoeve J, Kluter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006; 24: 1294-1301 |
| [31] | Rebelatto CK, Aguiar AM, Moretao MP, Senegaglia AC, Hansen P, Barchiki F, et al. Dissimilar differentiation of mesenchymal stem cells from bone marrow, umbilical cord blood, and adipose tissue, Exp Biol Med (Maywood) 2008; 233: 901-13 |