-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Polymer Science
p-ISSN: 2163-1344 e-ISSN: 2163-1352
2013; 3(2): 7-12
doi:10.5923/j.ajps.20130302.01
Structural-relaxation Mechanism of Glassy-like Polymers Plasticization
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOleg A. Fridman
Vladimir State University named after Alexander and Nikolay Stoletovs. Gorky street, 87, Vladimir city, 600000, Russian Federation
Correspondence to: Oleg A. Fridman, Vladimir State University named after Alexander and Nikolay Stoletovs. Gorky street, 87, Vladimir city, 600000, Russian Federation.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Structural and relaxation aspects of plasticization on the example of cellulose acetate plastics are studied. Three types of plasticization are examined: the plasticizer is miscible with the polymer at processing and operating temperatures; the plasticizer is miscible with the polymer at processing temperature and is immiscible at the operating temperature; the plasticizer is also immiscible at the operating temperature, but does not form the separate phase during molding. Plasticized material properties are determined not only by the manner of plasticizer effect on the material structure formation, but also by the extent of its influence on the relaxation rate at the test temperature.
Keywords: Plasticization, Cellulose Acetate, Miscibility of Components, Structure, Glass Transition Temperature, Relaxation, Properties
Cite this paper: Oleg A. Fridman, Structural-relaxation Mechanism of Glassy-like Polymers Plasticization, American Journal of Polymer Science, Vol. 3 No. 2, 2013, pp. 7-12. doi: 10.5923/j.ajps.20130302.01.
1. Introduction
- Development of polymeric material processing technology is aimed at the increase of equipment performance. Hence, the article molding time becomes comparable or exceeds that of polymeric material relaxation. Sometimes, this is the necessary condition for adhering required property, for instance, when fibers or oriented films are manufactured. However, relaxation time of a polymeric material should commonly be shorter than the article molding time.Plasticization is one of the methods reducing polymeric material equilibration time. Plasticizer must also perform this function in the finished product preserving its properties in temperature and mechanical stress ranges specified by operating conditions. It is noteworthy that complete separation of the plasticizer effect on pre-history of the sample and its behavior during experimental studies of plasticized polymer properties is hardly possible.Physicochemical and process aspects of plasticization of polymers are optimally discussed in monographs[1-5]. The authors of these works have noted that many experimental facts are not adequately explained. The works[6-7] contain the data regarding compatibility and properties of diacetate of cellulose plasticizied with diehylphthlate. The general theoretical framework for quantifying changes in the “high-frequency” relaxation dynamics of mixtures based on classical transition state theory has been developed[8]. The antiplasticization-to-plasticization transition phenomena was observed in high-frequency dielectric measurements. The phenomena of antiplasticization was investigated was observed in the work[9]. Oligomeric polycaprolactone was used for the modification of cellulose acetate by reactive processing in the works[10, 11]. Plasticization of cellulose diacetate by graft copolymerization of ε-caprolactone and lactic acid was investigated in work[12]. The thermal and mechanical properties of blends of poly(ε-caprolactone) with cellulose acetate in different proportions were investigated by differential scanning calorimetry and tensile property measurements[13].Structural and relaxation aspects of glassy polymer plasticization may be considered on the example of cellulose-acetate plastics. The subject of research is selected due to the following reasons:- cellulose acetate can only be processed into articles in the plasticized state;- cellulose-acetate plastics are operated under permanently varying stresses reasoned by the fact that articles from them are virtually exclusively used in combination with other materials (metal, glass, synthetic polymers), and when temperature changes, strains due to difference in thermal expansion coefficients occur.In the recent decade, the investigations were aimed at creation of biodegradable plastics from cellulose acetate[10-13]. This ultimate objective defined the plasticizer selection. In this work, we attempted to clear out general approaches to analysis of the plasticizing action of various types of plasticizers.
2. Experimental
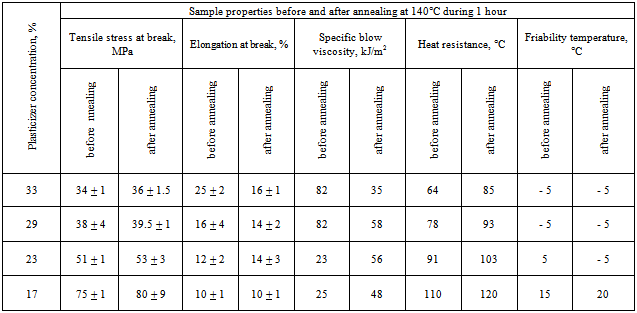
- The investigation was carried out on industrial production run of cellulose acetate which has hydroxyl group substitution degree 2.5 and average degree of polymerization 275. Plasticized compounds were produced by mixing components in a paddle mixer. Thermal plastication was carried out in a single-screw molding machine with screw diameter of 25 mm. Depending on the type and concentration of plasticizer the temperature in the course of extrusion varied to get melt viscosity of 103 - 104 Pa·sec. Test samples were produced with the molding machine providing 20 cm3 as maximum injection capacity and clamping force of 10 t. Total manufacturing cycle of one cast molding was 60 sec. The time of molten state is provided during 10 min. Films with thickness of 0.08 mm were prepared from 10% solution in acetone on the glass substrate.Glass transition temperature was determined according to thermo-mechanical curves made at permanent tensile stress equal 0.4 MPa and rate of temperature increase is 2 deg/min. The phase separation point refers to the plasticizer concentration at which the glass transition temperature of cellulose acetate stopped decreasing in spite of plasticizer concentration increases.Diethyl phthalate vapor pressure over plasticized cellulose acetate was measured at 50ºС by Knudsen effusion method. The pressure in the system during the experiment was (2-8) .10-4 Pa. Test samples were made in the form of films which were then triturated with the particles dimension of maximum 0.1 mm. The diethyl phthalate concentration at which minimum vapor pressure rate changes to pure plasticizer vapor pressure was referred to as the phase separation point.Capillary viscosimeter with capillary length of 12.4 mm and diameter of 1.6 mm was used to carry out rheological analysis. Vicat softening point was identified in liquid medium under the load of 10 N according to ISO 306. Friability temperature was assessed through analysis of srtrength-strain diagram (ISO R52) made at different temperatures in a tensile testing machine chamber. Friability temperature refers to the temperature at which brittle rupture was first observed with the test temperature decreases. Impact strength was assessed in unnotched samples according to the method of Sharpy (ISO 179). The flexural test measures the force required to bend a beam under three point loading conditions was determined according to ISO 178.
3. Results and Discussion
- Strictly speaking, a possibility of using a particular compound as a plasticizer may be assessed by composing a diagram of the polymer-plasticizer system state and correlation of phase equilibrium curve location with the range of plasticizer concentrations and article operating temperatures used in practice. Figure 1 shows experimental diagram of cellulose acetate – diethyl phthalate system state. The phase separation curve (1) is composed basing on results of differential scanning calorimetry method obtained at diethyl phthalate concentrations exceeding 60%[6]; by degeneration of cellulose acetate film glass transition temperature dependence on absorbed diethyl phthalate[7], and dependence on the composition of the polymer-plasticizer system with respect to diethyl phthalate vapor pressure at 50C[14].As observed in the Figure, the flow temperature determined as the temperature providing effective viscosity of 5•103 Pa•s[15] at the shear stress on the capillary viscosimeter wall equal 2.23•104 Pa lies intentionally above the separation curve, i.e. in the area of dynamic miscibility of cellulose acetate with diethyl phthalate. For obtained articles, the field of formulations and operating temperatures is limited by “heat resistance” parameter from above and friability temperature from beneath. These parameters, together with the flow temperature, also limit the range of plasticizer concentrations actually used in industry.
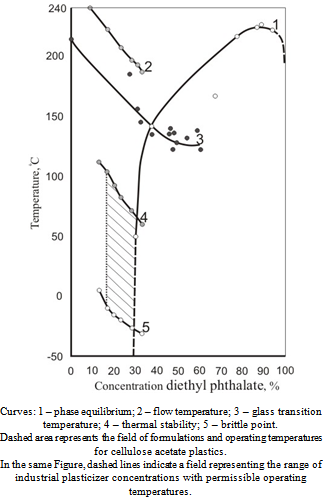 | Figure 1. The state diagram for cellulose acetate (with substitution degree of 2.33) – diethylphthalate system |
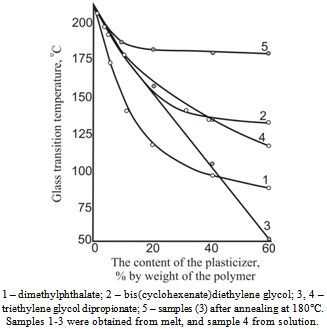 | Figure 2. Dependence of cellulose acetate glass transition temperature on plasticizer concentration and the injection method |
 | Figure 3. Dependence of cellulose acetate glass transition temperature on plasticizer concentration and the injection method |
|
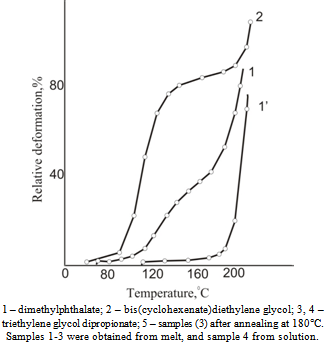
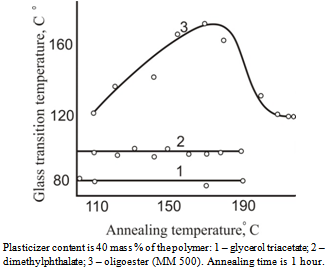 | Figure 4. Dependence of glass transition temperature on annealing temperature for cellulose acetate plastics |
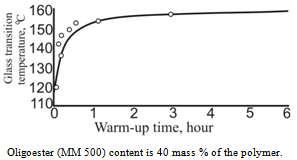 | Figure 5. Dependence of glass transition temperature on annealing time at 140C for cellulose acetate plastics |
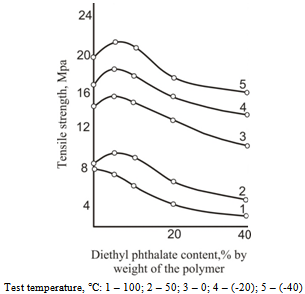 | Figure 6. Dependence of tensile stress at break on diethylphthalate concentration for cellulose acetate films |
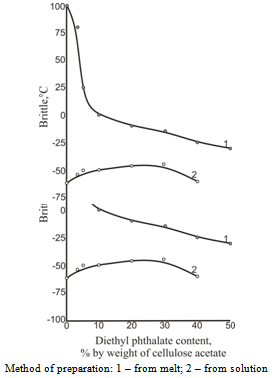 | Figure 7. Brittle temperature dependence on diethylphthalate concentration |
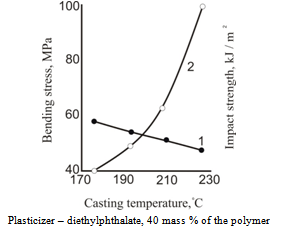 | Figure 8. Dependence of bending stress (1) and blow viscosity (2) on molding temperature for cellulose acetate plastic |
4. Conclusions
- It should be concluded that the three types of plasticization are existed: the plasticizer is miscible with the polymer at processing and operating temperatures; the plasticizer is miscible with the polymer at processing temperature and is immiscible at the operating temperature; the plasticizer is also immiscible at the operating temperature, but does not form the separate phase during injection molding.The reducing of the relaxation time for macromolecules, plasticization allows production of structurally homogeneous materials and, at the same time, promotes relaxation of stresses, which occur during operation of the article, i.e. provides deformation of polymeric material, in the given operation range without integrity break (without degradation). We believe that these are premises for stating definitions of “plasticizer” and “plasticization”.