-
Paper Information
- Next Paper
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Polymer Science
p-ISSN: 2163-1344 e-ISSN: 2163-1352
2012; 2(5): 91-101
doi: 10.5923/j.ajps.20120205.03
Emerging Issues in the Mechanisms of High Pressure Free Radical Ethylene Polymerization: A Review
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLKehinde A. J 1, Usman M. A 1, Edoga M. O 2, Owolabi R. U. 3
1University of Lagos, Chemical Engineering Department, Akoka, Lagos State, Nigeria
2Federal University of Technology, Chemical Engineering Department, Minna, Niger State, Nigeria
3Fountain University, Industrial and Environmental Chemistry Department P.M.B 4491, Osogbo Osun State, Nigeria
Correspondence to: Owolabi R. U. , Fountain University, Industrial and Environmental Chemistry Department P.M.B 4491, Osogbo Osun State, Nigeria.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The need to improve the performance of the high pressure polymer production process, and timely introduction of new and functionalised products even at reduced production cost which is becoming critical to staying in business has given room for overall studies of the ethylene polymerization process. In this review, emerging issues in the high pressure ethylene polymerization process through free radical approach is unveiled and presented. Different views and approaches of several authors on the tasking issues were compiled, analysed and discussed. Future researchable areas were made clear in this study. Further investigations were also made to model kinetically the high pressure ethylene polymerization reaction in tubular reactors only using mass balances and moment analysis. Although not discussed in this paper, the modeling of heterogeneous polymerization reactions such as precipitation polymerization and emulsion polymerization remains a challenge.
Keywords: Ethylene Polymerization, Free Radical, Tubular Reactors, Moment Analysis, Precipitation Polymerization, Emulsion Polymerization
Article Outline
1. Introduction
- The most important industrial reaction of ethylene is its reaction with itself (Polymerization)[1] . Polymerization as defined by Carothers[2] is the intermolecular combinations that are functionally capable of proceeding indefinitely. Polyethylene, the product of the indefinite combination of ethylene monomers at high pressure in the presence of initiators was the unexpected result of an experiment undertaken in the course of a fundamental research in the early 1930`s without a direct commercial target in view. Imperial Chemical Industries (ICI) in England decided to evaluate the effect of ultra-high pressures on some 50 chemical reactions. In 1933, an experiment was carried out, compressing ethylene gas to 1400 bar. A white solid was formed in the heavy steel vessel, which proved to be low-density polyethylene (LDPE). Subsequent work showed that minute traces of oxygen had caused the polymerization[3]. Polyethylene accounted for 30 % of the total annual world polymer consumption in 2007 (more than 70 million tonnes in 2007)[4] and are the most widely 70 million tonnes in 2007)[4] and are the most widely utilized synthetic polymers. Tubular process using a very long small diameter tubular reactor and an autoclave process using a well stirred tank reactor are commonly used industrial process for polyethylene synthesis. Hee-Jong Lee et al[5] confirmed that polyethylene products manufactured by these processes differ in their molecular architecture and end use properties. Majid[6] reported the scientific, industrial and commercial reasons for the enormous consumption, such as good chemical resistance, zero toxicity, bio-acceptability, good physical and mechanical properties, low cost, ease of fabrication, good raw material availability and low environmental impact[7-11]. From the S curve of polyethylene technology[12-13], it can be seen that polymerization processes caused revolutionary improvements in polyethylene technology as shown in Figure 1.In most textbooks and monographs on free radical ethylene polymerization, only very few if any at all commented on the challenges, emerging issues, unsolved problems and future opportunities of this part of macromolecular chemistry (Ethylene polymerization). Therefore, this mini review deals mainly with the identification of cloudy areas and gaps uncovered with a view of creating future researchable areas in free radical ethylene polymerization.
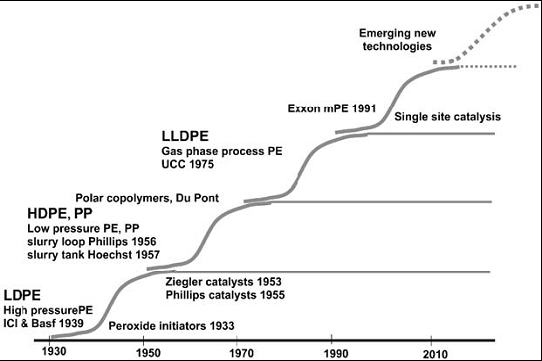 | Figure 1. S curve of polyethylene technology[12-13] |
2. Kinetics of Ethylene Polymerization
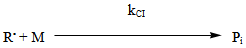
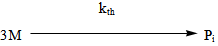
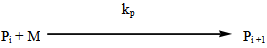
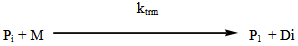
- The industrial importance of ethylene polymerization has led to extensive studies of its kinetics[14-17] . In this section, a set of reaction mechanisms required to model the kinetics of free radical polymerization of ethylene is described. A unique and unified set of elementary steps for free radical ethylene polymerization is still being debated and yet to be found in open literature. Buback,[18] studied the thermally initiated polymerization of ethylene. His results from the kinetic experiments on pure ethylene carried out at temperatures 180 -250℃ and pressures up to 2500 bars showed that a very slow thermally initiated reaction resulting in high molecular weight polyethylene could be established. The actual mechanism is not known but it can be written as an overall third order reaction. However, many workers in this field[14, 19-22] and recently by Kiparissides[23-24] proposed the kinetic mechanism described as follows;1. Initiation: (Activation) with Peroxides, Azo Compounds or Oxygen:
 | (1) |
 | (2) |
 | (3) |
 | (4) |
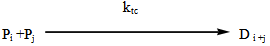 | (5) |
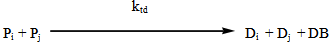 | (6) |
 | (7) |
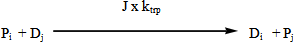 | (8) |
 | (9) |
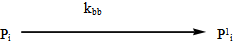 | (10) |
 | (11) |
3. Chemistry and Effectiveness of Initiators
- According to Zabisky[36] , Oxygen initiation of ethylene polymerization was first reported by Fawcett et al and has been used commercially in both tubular and batch reactors. It is well known that oxygen serves as an inhibitor in free radical polymerization at lower temperatures. Oxygen may react with radicals or monomers to form polymeric peroxides. These peroxides may then decompose to initiate the polymerization. Several authors including Thies and Schoenemann[37] have tried to model oxygen initiation by assuming an overall second order reaction of the type neglecting the inhibition reactions but with moderate success.
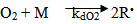 | (12) |
4. Kinetic Parameters and Estimation
- For each of the elementary steps stated in equation (1) to (11), three parameters were used to determine the value of their kinetic rate constant at any temperature and pressure according to the modified Arrhenius equation:
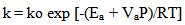 | (13) |
 | Figure 2. Schematic representation of the principle of heterofunctional initiators, more specifically dual initiators.[47] |
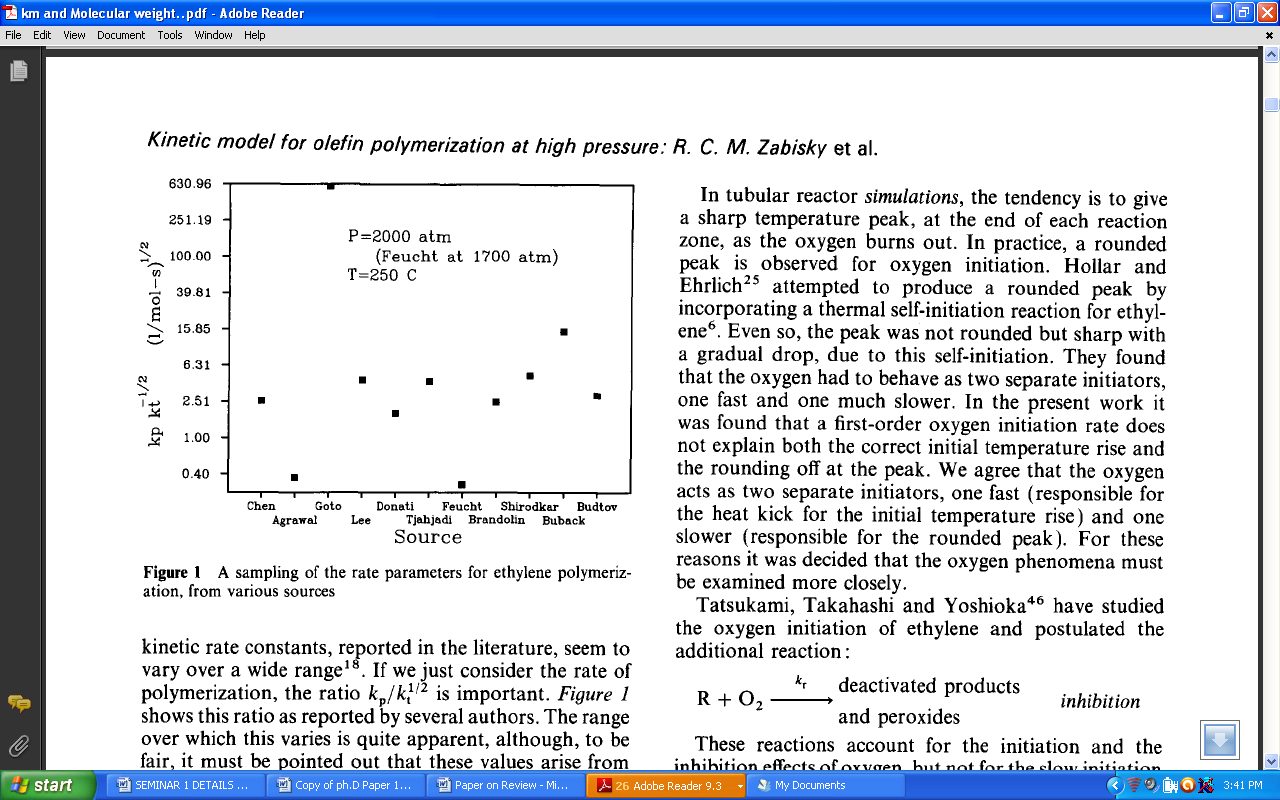 | Figure 3. A sampling of the rate parameters for ethylene polymerization from various sources.[36] |
|
5. Ethylene Conversion/Polymer Molecular Weight
- Literature search shows that common ethylene conversion obtained by various researchers falls within the range of 14 – 24 % as shown in Table 2.Dhib and Nidawy,[39] however observed that the thermodynamic conditions of the process hinder ethylene from going to full conversion. Other than recycling the product, one way of improving the monomer conversion is to initiate the polymerization with difunctional organic peroxides. Due to the dual functionality of difunctional peroxide, the ethylene conversion they obtained from their model was about twice as much as that obtained with monofunctional peroxide, for only a half amount of the initial initiator concentration. However, the setback of obtaining only limited low conversion of ethylene in high-pressure polymerization has been persistently an unpleasant and discouraging reality in spite of the classical approach to enhancing the conversion upon recycling the product. Another route of improving ethylene conversion in this kind of polymerization process is to investigate the effectiveness of initiators. Aside the conversion, the production of polymers with desired end-use properties is of significant financial importance to the polymer industry. One of the most important molecular properties that control the end-use characteristics of polymers is the molecular weight distribution (MWD) as it directly affects the physical, mechanical and rheological properties of the final product[55] . The molecular weight distribution of a polymer can be characterized by the number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity (PD). MWD/PDI is considered as a fundamental property that determines polymer properties and thus its applications. An ideal polymer contains only one type of polymer with the same architecture, microstructure and chain length as shown for the polymer chain composition I in Scheme 1[56].
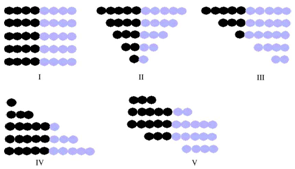 | Figure 4. Scheme 1. Different Polymer Chain Compositions |
6. Harsh Operating Conditions
- Large number of the now available polymers are obtained, on industrial scale, through high pressure process and any improvement of the industrial process has important economic consequences. In both tubular and autoclave reactors , a free radical mechanism using initiators such as peroxides or oxygen takes place at pressures ranging from 1,000 to 3000 atm[49]. Free radical polymerization of ethylene is carried out at high pressure and elevated temperature[57]. Temperatures exceeding 300℃ cause ethylene to decompose and are not recommended in practice. The high-pressure process is usually a bulk polymerization initiated by organic peroxide. The high pressures (>2000 bars) and temperatures (>250℃) make it difficult to obtain accurate kinetic constants using small scale laboratory experiments. Thus, one has to rely on published literature for kinetic data. Unfortunately, there is very little agreement between the various publications.Over the last few decades, a lengthy list of academicians and industrialists incessantly attempted to establish a unifed tangible understanding of ethylene polymerisation in high-pressure autoclave reactors[21, 58-60] and tubular reactors[28, 61] mainly because the technical properties of polymer products are mainly determined by the conditions of the polymerization reaction. Kiparissides et al.[49] reported that low pressure ionic ethylene polymerization processes have been developed for the production of high density polyethylene and medium density polyethylene.The process requires low pressure(8-80 atm) and temperatures less than 150 oC using a transition metal catalyst of the Ziegler-Natta or Phillip type. Though the process has gained a lot of popularity in the polyolefin industry still, there is the need for a strong reconsideration of polymerization processes of the most common monomers such as ethylene with emphasis on new initiating systems under low pressure and "uncatalyzed" polymerizations. Similarly, despite the commercial success of gas phase ethylene polymerization technology, the public literature contains no accounts of fundamental scale-up studies of gas phase processes. There is further need, therefore, for a comprehensive understanding of detailed polymerization behavior in gas phase polymerization. A challenge for academic researchers studying gas phase polymerization of ethylene is how to scale down commercial processes for experimental laboratory studies.
7. Ethylene Polymerization Kinetic Model Development for a Tubular Reactor
- According to Xie et al.[62] reactor modeling is the determination of a quantitative relationship between reactor performance and reactor operating conditions. It requires comprehensive understanding of polymerization processes, physical phenomena, and chemical reaction mechanisms. The importance and benefits of reactor modeling have been widely recognized by both industrial and academic researchers. The application capability of a model depends on scope of the modeling effort. Ray[63-64] defined a modeling hierarchy as microscale, mesoscale, and macroscale, according to the characteristics of the polymerization reactor systems. The relationships between modeling scale and polymerization systems are similarly outlined in Figure 5 after Ray[63-64]. The emphasis of each modeling level, in particular for ethylene polymerization systems, can be summarized as follows:1. Microscale modeling: Polymerization rate development, or polymer yield; reactant species distribution; molecular weight development and its distribution; chemical composition of polymer chains and its distribution; microstructures of polymer chains, including chain branching, unsaturated groups, and sequence distribution.2. Mesoscale modeling: Interphase heat and mass transfer; intraphase heat and mass transfer; fluid mechanics and micro-mixing; polymer particle morphology development; polymer particle size and distribution.3. Macroscale modeling: macro-mixing and residence time distribution; overall material and energy balance; heat and mass transfer from the reactor; reactor dynamics and control; polymer grade transition and control.
 | Figure 5. Levels of polymerization reactor modelling[63-64] |
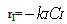 | (14) |
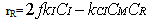 | (15) |
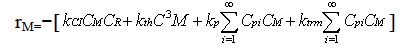 | (16) |
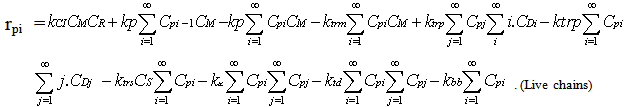 | (17) |
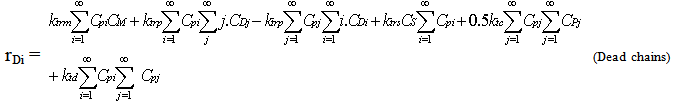 | (18) |
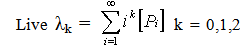 | (19) |
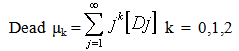 | (20) |
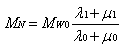 | (21) |
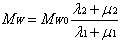 | (22) |
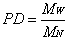 | (23) |
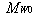 is the monomer molecular weight.Therefore,
is the monomer molecular weight.Therefore,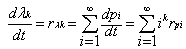 | (24) |
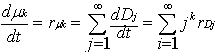 | (25) |
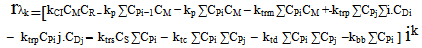 | (26) |
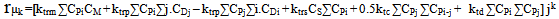 | (27) |
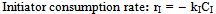 | (28) |
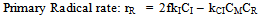 | (29) |
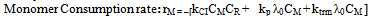 | (30) |
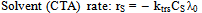 | (31) |
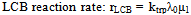 | (32) |
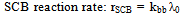 | (33) |
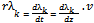 | (34) |
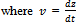 | (35) |
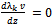 | (36) |
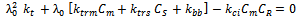 | (37) |
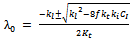 | (38) |
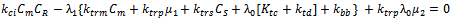 | (39) |
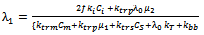 | (40) |
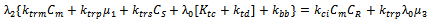 | (41) |
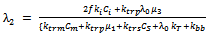 | (42) |
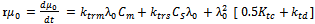 | (43) |
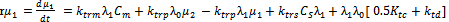 | (44) |
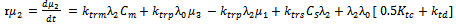 | (45) |
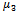 which would require an additional differential equation. Following the approach of several authors[36 ,39] , the closure of Hulburt and Katz[67] gives the following expression for
which would require an additional differential equation. Following the approach of several authors[36 ,39] , the closure of Hulburt and Katz[67] gives the following expression for 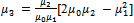 | (46) |
8. Conclusions
- In this review, some issues concerning the mechanism of ethylene polymerization, chemistry and effectiveness of initiators, ethylene conversion/polymer molecular weight, harsh operating conditions, model parameters estimation techniques, were discussed. The areas that need further research attention have been identified. One of the outstanding issues is to develop more efficient parameter estimation techniques to strictly limit the wide discrepancies in published data on ethylene polymerization kinetic parameters and the possibility of synthesizing polyethylene of improved quality under mild conditions. Kinetic modeling plays an important role in the design of polymerization reaction conditions with which to tailor a polymer’s molecular architecture. This area was not left out as attempt was also made to develop appropriate kinetic model for ethylene polymerization in a tubular reactor based on experimentally determined elementary steps found in the literature which could be used in estimating the first three moments of both the live and the dead radicals which are used to estimate number average molecular weight, weight average molecular weight and polymer polydispersity.SYMBOLS USEDCI Concentration of initiation, mol/LCM Concentration of monomer mol/LCR Concentration of free radical 1/SCS Concentration of solvent mol/LDi ead polymer or dead chainsf Efficiency of original initiation I Initiationkbb Rate constant of backbiting 1/SkI Rate constant of oxygen/peroxide initiation, 1/SkP Rate constant of propagation, 1/mol.Sktc Rate constant of termination by combination, 1/mol.Skth Rate constant of monomer thermal initiation, 1/Sktrm Rate constant of chain transfer to monomer, 1/mol.Lktrp Rate constant of chain transfer to polymer,1/mol.Sktrs Rate constant of chain transfer to solvent,1/Skβ Rate constant of β-scission to secondary radical.1/SLCB Long chain branchingM MonomerMwo Monomer molecular weight Pi Live radical, radical polymer, live polymer or live chainsPDI Polydispersity IndexR Initiator radicalS SolventSCB Short chain branchingλ Live polymer momentμ Dead polymer momentλ0 Zeroth live polymer momentλ1 First live polymer momentλ2 Second live polymer momentμ0 Zeroth dead polymer momentμ1 First dead polymer momentμ2 Second dead polymer momenti,j Ultimate monomer unit in the radical chain
References
| [1] | Mc Duell, G.R., (1984), A-level chemistry,Charlse Letts Ltd. |
| [2] | Carothers, W.H. (1931), Polymerization, tiber die angeblichen Isomerien bei cyclischen Oxalsaureester, Chem. Rev., 8, 353-426. |
| [3] | Charlie, E.S. (2006), Exxon Mobil High Pressure Process Technology for LDPE. |
| [4] | Scheidl, K. (2008), Polyethylene-Polypropylene Chain Global PE/PP Industry Report. Maack Business Services, MaacK/Scheidl Partnership, Plastics Technology and Marketing 28th Anniversary. |
| [5] | Hee, J.L, Yeong, K.Y and Jin,Y.C (2000), Modeling of Industrial High Pressure Autoclave Polyethylene Reactor Including Decomposition Phenomena, Korean J.Chem.Eng., 17 (2) 223-229. |
| [6] | Majid, D. (2009), Comparison of Catalytic Ethylene Polymerization In Slurry and Gas Phase, Ph..D Thesis, University of Twente,The Netherlands. |
| [7] | Frosch, R.A. and N.E. Gallopoulos, (1989) Strategies for Manufacturing. Scientific American, 261 (3) 144-152. |
| [8] | Galli, P. and G. Vecellio,(2001), Technology: driving force behind innovation and growth of polyolefins. Progress in Polymer Science, 26 (8): 1287- 1336. |
| [9] | Galli, P. and G. Vecellio, (2004), Polyolefins: The most promising large-volume materials for the 21st century. Journal of Polymer Science Part a-Polymer Chemistry, 42 (3) 396-415. |
| [10] | Tannous, K. and J.B.P. Soares,(2002) Gas-phase polymerization of ethylene using supported metallocene catalysts: Study of polymerization conditions. Macromolecular Chemistry and Physics, 203(13) 1895-1905. |
| [11] | Romano, U. and F. Garbassi, (2000), The environmental issue. A challenge for new generation polyolefins. Pure and Applied Chemistry, 72 (7) 1383-1388. |
| [12] | Knuuttila, H., Lehtinen, A and Nummila-Pakarinen, A (2004), Advanced polyethylene technologies - Controlled material properties. Long-Term Properties of Polyolefins, 169: 13-27. |
| [13] | Montagna, A.A., R.M. Burkhart, and A.H. Dekmezian, (1997),The evolution of single-site catalysis. Chemtech, 27 (12) 26-31. |
| [14] | Ehrlich, P. and Mortimer, G. A. (1970). Fundamentals of the Free-Radical Polymerization of Ethylene. Adv. Polym. Sci. 7: 386-448. |
| [15] | Zhang, S.X., Read, N.K., Ray, W.H., (1996), Runaway Phenomenom in Low Density Polyethylene Autoclave Reactor ,AICHE J., 42:2911-2925. |
| [16] | Fogler, H. S. (1999), Elements of Chemical Reaction Engineering. Prentice Hall International Series in the Physical and Chemical Engineering Sciences, 3ed. |
| [17] | Costas, P.B, Sundaram, R., John, F. (2001), Physical Properties, Reactor Modeling And Polymerization Kinetics In The Low Density Polyethylene Tubular Reactor Process, Ind.Eng.Chem.Res.41 (5), 1017- 1030. |
| [18] | Buback, M., (1980), The high pressure ethylene polymerization of pure ethylene, Makromol Chem.,18,373. |
| [19] | Luft,G.,Kampf,R.,Seidi,H., (1982),Angew.Makromol.Chem.,108,203. |
| [20] | Yamamoto,K.and Sugimoto.M.(1979), Rate Constants For Long Chain Branch Formation In Free Radical Polymerization of Ethylene,J.Macrom.Sci-chem., 13 (8),1067. |
| [21] | Goto, S., Yamamoto, K., Furui, S., and Sugimoto, M. (1981). Computer model for commercial high pressure polyethylene reactor based on elementary reaction rates obtained experimentally. Journal of Applied Polymer Science: AppliedPolymer Symposium, 36, 21–40. |
| [22] | Ogo,Y., (1984), Polymerization at High Pressures, JMS-Rev..Macrom.Chem.Phys., 24 (1),1 |
| [23] | Kiparissides, C. (2006). Challenges in Particulate Polymerization Reactor Modeling and Optimization:A Population Balance Perspective.Journal of Process Control 16,205. |
| [24] | Pladis, P., and Kiparissides, C. (1998). A Comprehensive Model For The Calculation of Molecular Weight-Long-Chain Branching Distribution in Free-Radical Polymerizations.Chemical Engineering Science, 53, 3315–3333. |
| [25] | Ham , J.Y. and Rhee, H.K, (1996) Modeling and Control of an LDPE Autoclave Reactor ,J. Proc.Cont., 6, 241. |
| [26] | Tsutomu, K., and Masatsugu, I(1968) J. Res.nst. Catalysis, Hokkaido Univ., Vol. 16, No.1, pp. 155 - 168 . |
| [27] | Agrawal, S. and Han, C.D (1975). AIChE J., 2 1 (3), 449. |
| [28] | Chen, C. H., Vermeychuk, J. G., Howell, J. A., and Ehrlich, P. (1976). Computer model for tubular high-pressure polyethylene reactors. AIChE Journal, 22 (3), 463–471. |
| [29] | Donati, G., L. Marini, G. Marziano, C. Mazzaferri, M. Spempinato, and E.Langianni (1982). Proc. 7th Int. Symp. CHem. Reaction Eng. (Boston), 579. |
| [30] | Lee, K.H. and J.P. Marano (1979). Am. Chem. Sot. Symp. 104, 221. |
| [31] | Luft, G. and H. Seidl (1981). J. Macromol. Sci-Chem., 415 (11) l-33. |
| [32] | Gupta, S. K., A. Kumar and M.V.G. Krishnamurthy (1985).Simulation of Tubular low density Polyethylene.Polym. Engr. Sci., 25 (1) 37-47. |
| [33] | Takahaski, T. and P. Ehrlich (1982). Macromolecules, 15, 714. |
| [34] | Nigam, K.M. and K.D.P. Nigam (1983). J. Appl. Polym. Sci., 281- 887. |
| [35] | Yoon, B.J. and H.K. Rhee (1985). Chem. Eng. Commum.. (34), 253. |
| [36] | Zabisky, R.C.M., Chan,W.M., Gloor, P.E.,and Hamielec ,A.E. (1992), A kinetic model for olefin polymerization in high pressure tubular reactors:a review and update,Polymer, vol.33,No.11,2243-2262. |
| [37] | Thies, J. and K. Schoenemann (1970). 1st Int. Symp. Chem. Reaction Eng,Washington, DC. |
| [38] | Kondratiev, J.N., Ivanchev, S.S., (2005), Possibilities For Optimization of Technological Modes For Ethylene Polymerization in Autoclave and Tubular Reactors,Chem. Eng Journal, 107, 221-226. |
| [39] | Dhib, N; Al-Nidawy, R; (2002), Modeling of Free Radical Polymerization Of Ethylene Using Difunctional Initiators, Chem.Eng.Sci, Vol.57,2735- 2746. |
| [40] | Luft, G., & Seidl, H. (1985). Application of bifunctional organic peroxides in the polymerisation of ethylene under high pressures. Die Angewandte Makromolekulare Chemie, 129, 61–70. |
| [41] | Luft, G., Bitsch, H., & Seidl, H. (1977). Effectiveness of organic peroxide initiators in the high-pressure polymerisation of ethylene. Journal of Macromolecular Science-Chemistry, 11(6), 1089–1112. |
| [42] | Seidl, H., and Luft, G. (1981). Peroxides as initiators for high-pressure polymerization. Journal of Macromolecular Science-Chemistry, 15(1), 1–33. |
| [43] | Luft, G., Lim, P., Pavlaskis, S., and Seidl, H. (1985). The decomposition of 2,2-bis(t- butylperoxide)butane under high pressure. Journal of Macromolecular Science-Chemistry, 22(9), 1183–1200. |
| [44] | Luft, G., and Dorn, M. (1988). Asymmetrical bifunctional organic peroxides as initiators for the high-pressure polymerisation of ethylene. Journal of Macromolecular Science-Chemistry, 25(8), 987–998. |
| [45] | Puts R.D and Sogah D.Y. (1997). Universal multifunctional initiator containing orthogonal reactive sites. Synthesis of macromonomers and comb polymers using consecutive controlled free radical and cationic ring-opening polymerizations. Macromolecules ; 30 (23) 7050–7055. |
| [46] | Hawker ,C.J, Hedrick, J.L, Malmstro, E.E, Trollsas M, Mecerreyes D, and Moineau G, (1998) Dual living free radical and ring opening polymerizations from a double-headed initiator. Macromolecules; 31 (2) 213–9. |
| [47] | Bernaerts ,K.V, Willet N, Van Camp W, Je´roˆme R, Du Prez FE. (2006) pH-responsive diblock copolymers prepared by the dual initiator strategy. Macromolecules ; 39: 3760–3769. |
| [48] | Matthew, J.S. (2005), Experimental and Modelling Investigation of a Novel Tetrafunctional Initiator In Free Radical Polymerization,Ph.D Thesis University of Waterloo,Canada. |
| [49] | Kiparissides, C., Verros, G., and Mcgregor, J. (1993). Mathematical Modeling, Optimization and Quality Control of High-Pressure Ethylene Polymerization Reactors. Journal of Macromolecular Science—Reviews in Macromolecular Chemistry and physics, 33: 437–527. |
| [50] | Brandolin, A.,Capiati, N.J.,Farber, J.N.,Vales, E.M.,(1988), Mathematical model for high pressure tubular reactor for ethylene polymerization,I.&E.C.Res. 27 (5) 784-790. |
| [51] | Fred, Z.Y., Ali, L., Simant, R.U, Ramdhane, D., (2004), Modeling ,Simulation and Optimal Control of Ethylene Polymerization in Non-Isothermal High Pressure Tubular Reactors, IJCRE,2 (16). |
| [52] | Zhou, W., Marshall, E., Oshinowo, L. (2001) Modeling LDPE Tubular and Autoclave Reactor,I.&E.C.Res., 40:5533-5542. |
| [53] | Brandolin,A.,Lacunza,M.H.,Ugrin,P.E.,Capiati,N.J.(1996),High pressure polymerization of ethylene.An improved mathematical model for industrial tubular reactor,Poly.Reac.Eng.4 (4),193-240. |
| [54] | Mavridis, H., Kiparissides, C. Optimisation of High Pressure Polyethylene Tubular Reactor, Pol.Proc.Eng., 3 (3), 263-290, 1985. |
| [55] | Verros, G.D. (2003), Calculation of molecular weight distribution in non-linear free radical copolymerization, Polymer 44 7021–7032. |
| [56] | Bernd, G. (2006), Molecular weight distribution in living polymerization, Progress in Organic Coatings ,55 ,189–193. |
| [57] | Odian, G., Principles of Polymerization,pp. 198–349. Wiley, 4th ed., 2004. |
| [58] | Feucht, P., Tilger, B., & Luft, G. (1985). Prediction of molar mass distribution, number and weight average degree of polymerisation and branching of low density polyethylene. Chemical Engineering Science, 40 (10), 1935–1942. |
| [59] | Lorenzini, P., Pons, M., and Villermaux, J. (1992a). Free-radical polymerization engineering-III. Modelling homogeneous polymerisation of ethylene: Mathematical model and new method for obtaining molecular weight distribution.Chemical Engineering Science, 47(15-16), 3969–3980. |
| [60] | Chan, W. M., Gloor, P. E., & Hamielec, A. E. (1993). A kinetic model for olefin polymerisation in high-pressure autoclave reactors. AIChE Journal, 39 (1), 111–126. |
| [61] | Shirodkar, P. P. and Tsien, G. O. (1986). A mathematical model for the production of low density polyethylene in a tubular reactor. Chemical Engineering Science, 41(4), 1031–1038. |
| [62] | Xie, T, McAuley, K,B , HSU, J,C and Bacon, D.W, Gas Phase Ethylene Polymerization: Production Processes, Polymer Properties, and Reactor Modeling, Ind. Eng. Chem. Res. 1994,33, 449-479. |
| [63] | Ray, W. H. (1986) . Modelling of Polymerization Phenomena. Ber. Bunsen- Ges. Phys. Chem. 90,947 |
| [64] | Ray, W. H.(1991) . Modelling of Addition Polymerization Processes-Free Radical, Ionic, Group Transfer, and Ziegler-Natta Kinetics. Can. J. Chem. Eng. 69, 626. |
| [65] | Arriola, D.J. (1989), Ph.D. Thesis ,Department of Chemical Engineering,University of Wisconsin,Madison. |
| [66] | Metzler, D.E., Harris, C.M., Johnson, R.A., Siano, D.B., Thomson, J.A., (1973), Spectra of 3- Hydroxypyridines: Brand Shape Analysis and Evaluation of Tautomeric Equilibria, Biochem., (12) 26,5377-5392. |
| [67] | Hulburt, H.M., Katz,S., (1964) ,Some Problems In Particle Technology: A Statistical Mechanical Formulation,Chem.Eng.Sci., 19: 555-574. |
| [68] | Bernaerts, K.V, Schacht, E.H, Goethals, E.J, Du Prez, F.E. (2003) Synthesis of poly(tetrahydrofuran)-b-polystyrene block copolymers from dual initiators for cationic ring-opening polymerization and atom transfer radical polymerization.J Polym Sci Polym Chem ;41(21):3206–17. |