-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2018; 8(1): 13-15
doi:10.5923/j.ajoc.20180801.03

Strategy for the Construction of Lactones via Ene-reaction
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLBello Y. Makama
Department of Maths and Science, American University of Afghanistan, Darulaman Road, Kabul, Afghanistan
Correspondence to: Bello Y. Makama, Department of Maths and Science, American University of Afghanistan, Darulaman Road, Kabul, Afghanistan.
| Email: |  |
Copyright © 2018 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

An effective ene-reaction protocol was developed for the preparation of cis-fused bicyclic lactones catalyzed by various Lewis acids at low temperatures.
Keywords: Ene-reaction, Lactones, Lewis acid, Low temperature
Cite this paper: Bello Y. Makama, Strategy for the Construction of Lactones via Ene-reaction, American Journal of Organic Chemistry, Vol. 8 No. 1, 2018, pp. 13-15. doi: 10.5923/j.ajoc.20180801.03.
Article Outline
1. Introduction
- The synthesis of the cis-fused bicyclic lactones relies extensively on the Lewis acid catalysed carbonyl-ene reaction which has been widely used to access carbocycles.1 In 1943 Alder discovered the ene-reaction and classified it in his Nobel Lecture as an “indirect substitution addition” or “ene synthesis" in 1950. The synthesis of the bicyclic lactone intermediate relies on the intramolecular ene reaction, a conversion that has been widely used as a tool to access polycycles. The conversion involves the reaction between an alkene having an allylic hydrogen (an"ene") and a compound containing an electron deficient double bond (an "enophile") to form a σ-bond with migration of the ene double bond and a 1,5-hydrogen shift Scheme 1.0 [1]
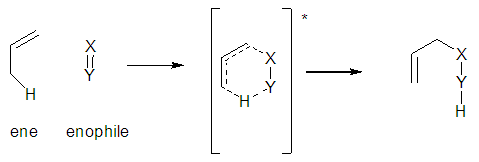 | Scheme 1.0 |
 | Scheme 1.1 |
2. Results & Discussion
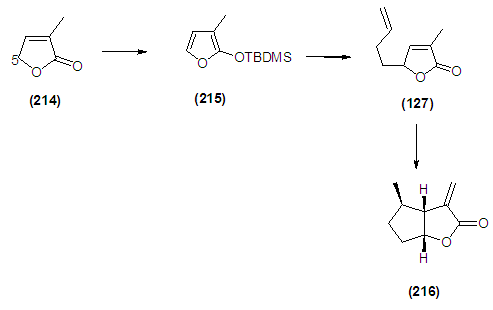
- The synthesis on a suitable scale of the key bicyclic lactone (216) would require an efficient and robust synthesis of the precursor (127). Initial inspection of the intermediate (127) suggests that it could be conveniently assembled by coupling the C-5 cabanion of 3-methyl-2(5H)-furanone (214) with the terminal cation of 4-bromobutene in a 1,4 addition protocol. Once this is achieved, subsequent modification is required, which involves the cleavage of the 1,3-dioxalane to aldehyde and a further ene-reaction protocol would afford bicyclic lactone (216). Unfortunately, the behaviour of 2-furanolate ions is inconsistent. Pattenden [2] has observed that the lithium enolate obtained from 3-methyl-2(5H)-furanone (214) on alkylation with various ally bromides gives both C-3 and C-5 substitutions. It was envisaged that C-5 regioselectivity could be induced by recourse to the 2-(trialkylsiloxy) furan, which behaves as the equivalent of the localized C-5 of the 2(5H)-furanone entity (214). Protection of the carbonyl group of 3-methyl-2-(5H)-furanone (214) was achieved following the procedure reported by Jefford. [3] It was envisaged that compound (215) could conveniently furnish compound (127) with high regioselectivity. The possibility that (215) could be converted to (127) was considered on the basis of a report by Jefford3 that ω-bromogeranyl acetate could undergo alkylation at C-5 position of siloxyfurans to furnish the corresponding butenolides in good yields. This model compound was considered to explore the efficacy of this transformation. It was envisaged that activation with silver salt could provide the desired selectivity. Having the essential reagents in hand, the next step was set out to put them together to create the precursor (127). The coupling was achieved with a stiochiometric amount of silver trifluoroacetate and, in keeping with previous findings of Jefford, alkylation occurred with high regioselectivity. Unfortunately in our systems the yield was found to be 9% for, when 4-bromobutene was used as the coupling agent. In an attempt to establish optimized conditions for this reaction, it was discovered the yield could be greatly increased to 69% for (127), when the temperature was kept at -78°C during the addition of 4-iodobutene (238) Scheme 1.2.
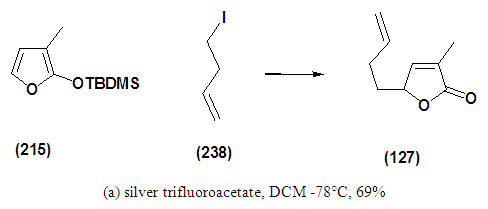 | Scheme 1.2 |
3. Lewis Acid Catalyze Carbonyl Ene-reaction
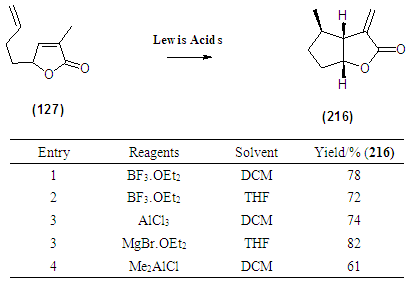
- The attempted synthesis of the cis-fused bicyclic lactone (216) and (217) was to involve the reactions of (127) using an ene-reaction using procedure reported by Snider. [4] According to this method, Me2AlCl was added to solutions of (127) in DCM at -78°C under nitrogen and the resulting mixtures were allowed to warm to room temperature with stirring for 48 hours. After workup, 1H NMR analysis revealed that (216) and (217) have been formed. The protocol was repeated with different Lewis acids to isolate the endo-diastreoisomer (216) in good yield. The stereochemical assignment of -cis at ring junction and the endo methyl group were achieved via nOe.
|
4. Conclusions
- Our proposed synthetic approach to the bicyclic lactone system systems using the ene-reaction a novel method was successful and gave the correctly placed lactones. It was deduced that it may be successful due to the Lewis acid complexing with the furan alkene, therefore, lowering the energy of the LUMO which affects the HOMO-LUMO interaction and allowed the cyclization protocol.