-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2018; 8(1): 1-7
doi:10.5923/j.ajoc.20180801.01

Investigation of an Amidation Reaction for the Preparation of Some 2-(2-Phenyl-4-Oxoquinazolin-3(4H)-yl)-N-Substituted Acetamides and N-Substituted 2-(2-(4-Oxo-2-Phenylquinazolin-3(4H)-yl)Acetylhydrazine-1-Carboxamides
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLOmaima M. Aboul Wafa1, Hoda M. G. Daabees2, Alaa A. El-Tombary1, Eman S. Ezz-El Dien2
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt
2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Damanhour, Damanhour, Egypt
Correspondence to: Omaima M. Aboul Wafa, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt.
| Email: |  |
Copyright © 2018 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A previously reported amide bond formation performed by treating ethyl 2-phenyl-4-oxo-3(4H)quinazolineacetate with the appropriate amine in boiling HOAc to synthesize a series of quinazolin-4-one derivatives was investigated. Elucidation of the structure of the synthesized products by IR, 1H NMR and 13C NMR indicated that, unfortunately, no amide bond was formed. The only product produced in all reactions was 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid even when using different amines. Repeating the afore-mentioned reaction, amide formation was attempted for the synthesis of a series of 4-oxoquinazolinecarboxamide derivatives by treating 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetyl)hydrazine carboxylate with the appropriate 1° or 2° amine in boiling HOAc. Unfortunately, the reaction did not proceed as designed and recovery of the starting ester was the only product detected. Structure elucidation of the product obtained was also made by IR, 1H NMR and 13C NMR spectroscopy which indicated that no amide bond was formed. Therefore, in the present investigation, N-substituted acetamide derivatives were synthesized, by using known synthetic approaches, from the corresponding acid via treatment with SOCl2 in different solvent systems followed by amidation with the selected amines.
Keywords: Quinazolin-4-ones, Amide formation, 2-phenyl-4-oxo-3(4H)quinazolineacetic acid, N-substituted acetamides, Ethyl 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetyl)hydrazinecarboxylate, Acetyl-N-phenylhydrazinecarboxamide
Cite this paper: Omaima M. Aboul Wafa, Hoda M. G. Daabees, Alaa A. El-Tombary, Eman S. Ezz-El Dien, Investigation of an Amidation Reaction for the Preparation of Some 2-(2-Phenyl-4-Oxoquinazolin-3(4H)-yl)-N-Substituted Acetamides and N-Substituted 2-(2-(4-Oxo-2-Phenylquinazolin-3(4H)-yl)Acetylhydrazine-1-Carboxamides, American Journal of Organic Chemistry, Vol. 8 No. 1, 2018, pp. 1-7. doi: 10.5923/j.ajoc.20180801.01.
Article Outline
1. Introduction
- Quinazolinone represents one of the most important classes of heterocycles possessing wide spectrum of biological activities [1] that has inspired medicinal chemists to introduce many bioactive moieties to this nucleus to synthesize new potential medicinal agents [2]. Coupling reactions between acid chlorides, esters or acids and substituted amines have been extensively studied for the synthesis of a huge number of amides [3-6]. In the present investigation, N-substituted acetamide derivatives 4a-f were synthesized by using known synthetic approaches. As far as acid chlorides are concerned as starting materials for amide formation, they can be easily obtained from the corresponding acids via treatment with SOCl2 in different solvent systems followed by reaction with the selected amines [3, 4]. Other coupling methods were also found using different reaction conditions [6].Applying one of the reported studies to synthesize series of quinazolin-4-one derivatives by treating synthetically available esters 3 and 6 with the appropriate amine in boiling HOAc was carried out as depicted in scheme 1. All synthesized compounds were characterized by elemental analyses, IR, 1H NMR and 13C NMR spectroscopy. Aiming at improving the yield of the reaction and purity of the product, it was required to reinvestigate the previously reported amide bond formation reaction using different reaction conditions.
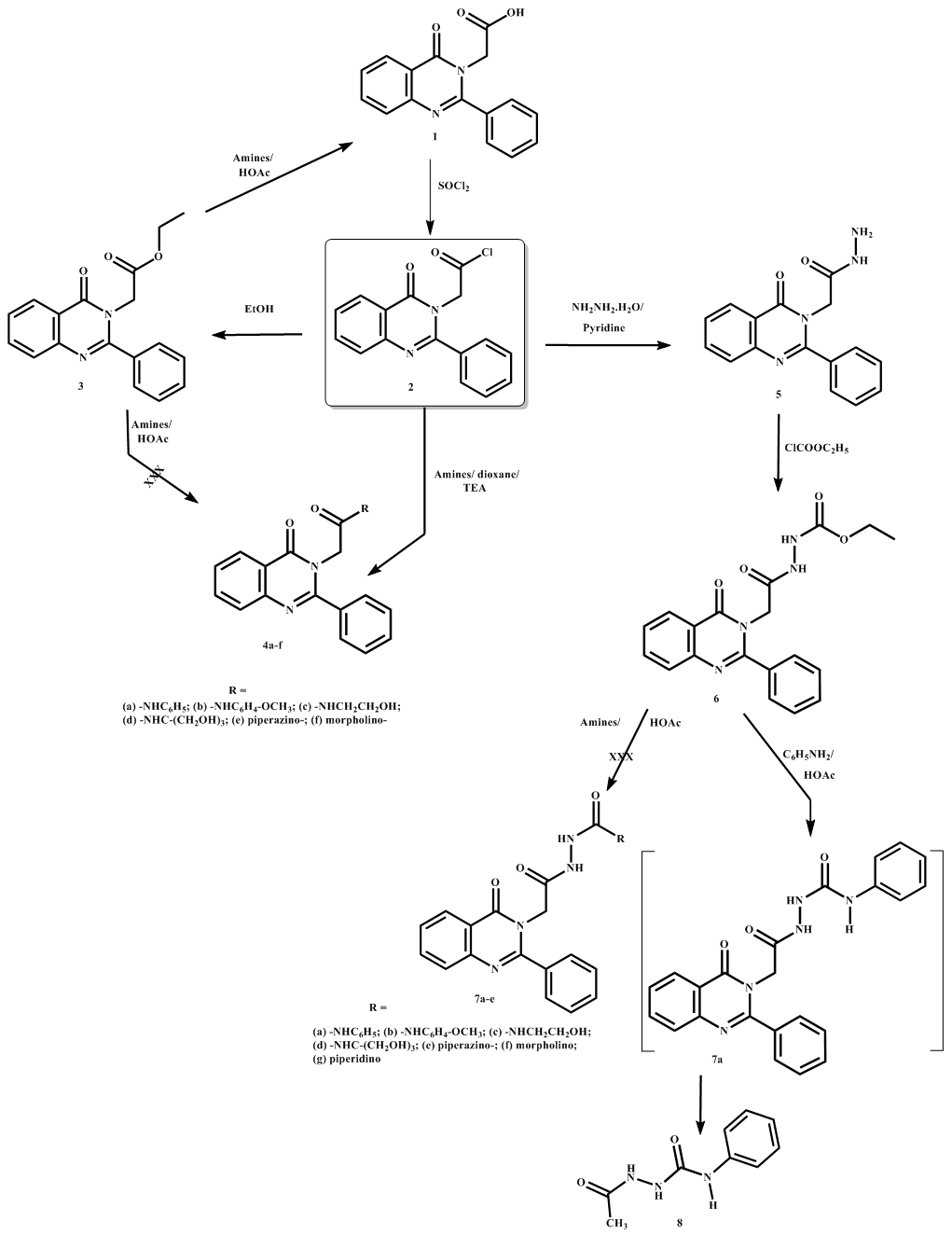 | Scheme 1. Synthesis of 2-(2-phenyl-4-oxoquinazolin-3(4H)-yl)-N-substituted Acetamides and N-substituted 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetylhydrazine-1-carboxamides |
2. Results and Discussion
- In the present work, attempt was made to prepare a series of 2-(2-phenyl-4-oxoquinazolin-3(4H)-yl)-N-substituted acetamides. Elucidation of the structure of the synthesized products by IR, 1H NMR and 13C NMR indicated that, unfortunately, no amide bond was formed and, consequently, products 4a-f were not obtained. The only product produced in all trials was 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid 1 [7] even when using differnt amines as aniline, p-anisidine, ethanolamine, tris-(hydroxymethyl)methylamine, piperazine and morpholine. Therefore, it was assumed that under the highly acidic conditions used, that is refluxing HOAc, hydrolysis of the ester has occured. IR spectra of the product, obtained from all reactions, showed stretching absorption bands for the carboxylic OH at high frequency. In addition, absorption bands for two C=Os, C=N, C=C Ar functionalities were also observed. Evidence was obtained by the superposition of IR spectra obtained for products obtained from reaction of the ester 3 with p-anisidine and ethanolamine. Similarily, 1H NMR and 13C NMR confirmed the proposed structure as 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid 1 for all reactions attempted. Comparison of the 1H NMR and 13C NMR spectra of product 1 obtained from both reactions, that is reaction of the ester 3 with ethanolamine and piperazine, showed a sharp D2O-exchangeable singlet at 12.20 ppm assigned for COOH. Quinazoline C7-, C6-, C8- and C5-protons resonated as 2 triplets and 2 doublets successively at their expected chemical shifts while CH2 protons resonated as singlet at 7.65 ppm. The 5 aromatic protons of 2-phenylquinazoline resonated as a downfield multiplet at its expected chemical shift. In 13C NMR of 1, chemical shift assignments were made for all 16 carbons in the molecule. 3ary and 4ary Carbons of the quinazoline nucleus, methylene-C and C2-phenyl ring were assigned downfield and were well resolved. The spectrum indicated the presence of two carbonyl carbons as evidenced by the appearance of 2 highly deshielded signals at 165.16 and 170.50 ppm assigned for C4=O and COOH respectively. Therefore, another method was adopted and conversion of 2-phenyl-4-oxo-3(4H)quinazoline acetic acid 1 with SOCl2 into the corresponding acid chloride derivative 2 was performed as the first step for amide formation. The acid chloride 2 was then used without further purification for amide formation by treatment with the appropriate amine as aniline, p-anisidine, ethanolamine, tri-(hydroxymethyl)-methylamine, piperazine, morpholine, in dry dioxane containing NEt3 [3]. Confirmatory evidence for structures 4a-f was provided by elemental analyses and spectral data (IR, 1H NMR and 13C NMR). IR spectra of compounds 4a-f showed stretching absorption bands for NH at high frequency. In addition, absorption bands for 2 x C=O, C=N, C=C Ar functionalities were also observed, υas and υs C-O-C stretching absorption bands were identified for compound 4b while OH stretching absorption bands were observed for compounds 4c,d. 1H NMR spectrum of 4a showed a downfield D2O-exchangeable singlet assigned for NH of the acetamide moiety. The usual singlet signal assigned for CH2 protons was included within multiplet assigned for aromatic protons which were detected at their expected chemical shifts. 1H NMR spectrum of 4b showed an upfield singlet assigned for OCH3 protons and a downfield D2O-exchangeable singlet assigned for NH of the acetamide moiety. Protons characterizing the p-methoxyphenyl group were detected as two well defined doublets. In 13C NMR spectrum of 4b chemical shift assignments were made for all 23 carbons in the molecule. 3ary and 4ary Carbons of the quinazoline nucleus, p-anisidine and C2-phenyl ring were assigned downfield and were well resolved. A shielded signal assigned for OCH3 and resonating at 55.68 ppm confirmed the compound structure. The two carbonyl carbons of side chain and the quinazolinone nucleus were assigned successively at 165.59 and 167.63 ppm. 1H NMR spectrum of 4c showed 2 upfield singlets resonating at 3.39 and 3.55 ppm assigned for NHCH2 and OHCH2 protons respectively and two downfield D2O-exchangeable singlets at 8.86 and 12.63 ppm assigned for NH of the acetamide moiety and OH respectively. The characteristic singlet signal assigned for CH2 protons was well detected at 4.78 ppm. 1H NMR spectrum of 4d showed an upfield singlet resonating at 4.45 ppm integrated for 6 protons and assigned for 3 x OHCH2 protons, well resolved after D2O, and two downfield D2O-exchangeable singlets at 8.07 and 14.83 ppm assigned for NH of the acetamide moiety and OH respectively. 1H NMR spectrum of 4e showed an upfield singlet resonating at 3.23 ppm, well resolved into 2 singlets at 2.96 and 3.55 ppm after D2O, integrated for 8 protons and assigned for 4 x CH2 protons of the piperazine moiety and a downfield D2O-exchangeable singlet at 14.24 ppm assigned for NH of the piperazine ring. In 13C NMR spectrum of 4e chemical shift assignments were made for all 20 carbons in the molecule. 3ary and 4ary Carbons of the quinazoline nucleus and C2-phenyl ring were assigned downfield and were well resolved. In addition, high field signals for the methylene carbons of the piperazine ring were also detected upfield at 66.72 ppm. A signal at 122.94 ppm was assigned for CH2CO while a highly deshielded signal at 165.20 ppm was assigned for the 2 carbonyl carbons of the molecule. 1H NMR spectrum of 4f showed two upfield singlets resonating at 3.15 and 3.82 ppm, integrated for 8 protons and assigned for 2 x NCH2 and 2 x OCH2 protons respectively of the morpholine moiety. Obviously, C7-H signal resonated at 7.06 ppm and split into ddd (doublet of doublet of doublet) with coupling constants of 1.08, 1.04, 0.60, 7.28, 7.68 Hz. The 2 J constants of 7.28 and 7.68 Hz belong to 2 different o-protons in the neighboring while J constants of 1.08, 1.04 Hz indicated the presence of am-proton. A similar pattern was observed for C6-H. Such pattern disappeared upon deuteration where 2 triplets were detected for C7-H and C6-H at 7.07 and 7.41 ppm respectively with again 2 different coupling constants for each of them. C8-H and C5-H signals resonated at their expected chemical shifts as 2 dd pattern changing into only 2 d upon deuteration. It was also noted that C2,6-Hs of the C-2 phenyl ring resonated as a dd at 8.04 ppm with 2 different coupling constants of 1.76 Hz for p- coupling and 7.92 Hz for o- coupling becoming a d after D2O at 7.98 ppm with J = 7.24 Hz. Increasing dilution of the sample after addition of D2O could be an explanantion for such change. Expanding the amidation reaction in HOAc, it was attempted to synthesize a series of quinazolinone derivatives by treating the ester 6 with the appropriate 1° or 2° amine, namely aniline, p-anisidine, ethanolamine, tri-(hydroxymethyl)-methylamine, piperidine, morpholine in boiling HOAc. Unfortunately, the reaction did not proceed as designed and recovery of the starting ester 6 was the only product detected. Structure elucidation of the product obtained was made by IR, 1H NMR and 13C NMR spectra which indicated that, unfortunately, no amide bond was formed and that the only product recovered in all trials was ethyl 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetyl)-hydrazine carboxylate 6 even when using differnt 1° and 2° amines. IR spectra of the product, obtained from all reactions, showed 2 stretching absorption bands for NHs at high frequency. In addition, stretching absorption bands for 3 x C=O (ester, quinazolinone and acetyl side chain) and υas and υs C-O-C functionalities were detected. Mixed vibrational bands for C=N and C=C Ar, were also observed. This was evidenced by the superposition of IR spectra obtained for products obtained from reaction of the ester 6 with tri-(hydroxymethyl)methyl amine and with piperidine. Similarily, 1H NMR and 13C NMR confirmed the recovery of the starting ester 6 for all reactions attempted. Study of the 1H NMR and 13C NMR spectra of the product obtained from the 2 reactions, that is reaction of the ester 6 with tri-(hydroxymethyl)methyl amine and with piperidine, are given for comparison purpose. 1H NMR spectra showed two D2O-exchangeable singlets resonating at low field assigned for the deshielded CONH and NHCOOEt protons respectively. The spectrum also showed a triplet and quartet assigned for CH3CH2 protons of the ester group at high field. In addition, CH2 and aromatic protons resonated downfield at their expected chemical shifts. In 13C NMR spectrum of compound 6, chemical shift assignments were made for all 19 carbons of the molecule. 3ary and 4ary Carbons of the phenyl ring and of the quinazoline nucleus were assigned downfield at their expected chemical shifts in addition to the high field signals due to methyl and methylene carbons of side chain ester. Three highly deshielded signals at 155.92, 156.90 and 160.27 ppm were assigned for the ester carbonyl and 2 carbonyl carbons of the quinazolinone nucleus and side chain, respectively. However, in only one case, attempt formation of N-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetylhydrazine-1-carboxamide 7a by treating ester 6 with aniline in refluxing HOAc according to the previously reported method produced the required amide 7a followed by instantaneous splitting of the side chain to afford 1-acetyl-4-phenylsemicarbazide 8 that had a melting point concordant with the one previously prepared [8]. Search in literature indicated that it was previously synthesized by treatment of N-acetylhydrazine with phenyl isothiocyanate in hot benzene [8]. IR, 1H NMR and 13C NMR confirmed the proposed structure as 1-acetyl-4-phenylsemicarbazide 8 for the reaction product. 1H NMR spectrum of 8 showed a D2O-exchangeable singlet at 9.92 ppm assigned for NHs. Quinazoline C7-, C6-, C8- and C5-protons and the 5 aromatic protons of 2-phenyl moiety were not identified at their expected chemical shifts. Instead, a singlet signal resonating at 2.04 ppm and integrated for 3 protons was assigned for the CH3 group. Two Downfield triplets at 7.02 and 7.28 ppm integrated for one and 2 protons were identified and assigned for C4-H and C3,5-Hs respectively. C2,6-Hs resonated as a doublet at 7.58 ppm. In 13C NMR spectrum of compound 8, chemical shift assignments were made for the 9 carbons present in the molecule. Again, downfield 3ary and 4ary carbons of the quinazoline nucleus and C2-phenyl ring could not be detected and were completely absent. The spectrum indicated that cleavage of the side chain has occurred as evidenced by the appearance of an upfield signal resonating at 24.45 ppm assigned for CH3 carbon and other deshielded signals assigned for the 6 carbon atoms of the phenyl ring of acetyl-N-phenylhydrazinecarboxamide 8. C=O carbons were highly deshielded and resonated at 168.72 ppm.
3. Experimental Section
- Chemicals used in the present investigation were purchased from Sigma-Aldrich Chemicals company, Inc., USA, and used without further purification. TLC was performed on aluminum sheets of silica gel 60F254, E-Merk, Germany, using iodine as visualizing agent. Melting points were determined in open-glass capillaries on an electro thermal melting point apparatus (Stuart Scientific, Model SMP1, UK) and were uncorrected. IR Spectra were recorded as KBr pellets on Shimadzu Infrared Spectrophotometer IR 470 at the Faculty of Pharmacy, Cairo University and Bruker Tensor 37 FT-IR Spectrometer, Central Laboratory Unit, Faculty of Science, Cairo University, Egypt and reported in cm-1. All 1H and 13C NMR spectra were recorded on Bruker Avance III apparatus 400MHz; Central Laboratory Unit, Faculty of Pharmacy, Cairo University, 400 MHz for 1H NMR and 100.63 MHz for 13C NMR and Varian Mercury VX 300 BB spectrophotometer; Faculty of Science, Cairo University, Egypt, 300 MHz for 1H NMR and 75.45 MHz for 13C NMR. Elemental analyses were run on Vario EL III German CHN Elemental analyser model, Regional Centre for Mycology and Biotechnology, Al-Azhar University, Nasr City, Cairo, Egypt.Attempt preparation of 2-(2-phenyl-4-oxoquinazolin-3(4H)-yl)-N-substituted acetamides from ester 3: Formation of 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid 1:Glacial acetic acid (5 mL) was added slowly while shaking to a mixture of ethyl (2-phenyl-4-oxoquinazolin-3(4H)-yl)acetate 3 (0.306 g, 1 mmol) and the corresponding 1° or 2° amine (1 mmol). The mixture was heated under reflux for 4-6 h, left to cool to R.T. and poured onto crushed ice. The obtained solid was filtered, dried and crystallized from EtOH. All products obtained had superimposable IR, 1H NMR and 13C NMR. The structure was elucidated and found to be 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid 1 [7]. M.p.: 133°C (Reported M.p.:130-132°C) [7]. IR (KBr, ύ, cm-1): 2924 (OH), 1686, 1643 (C=O), 1609 (C=N), 1590, 1497 (C=C Ar). 1HNMR (DMSO-d6, 400 MHz, δ ppm): 7.22 (t, 1H, C7-H, J = 7.32 Hz), 7.60 (t, 1H,C6-H, J = 7.6 Hz), 7.65 (s, 2H, CH2), 7.66-7.69 (m, 3H, C3',4',5'-Hs), 7.96 (d, 2H,C2',6'-Hs, J = 8.3 Hz), 8.07 (dd, 1H, C8-H, J = 8.3 Hz, 1.36 Hz), 8.73 (d, 2H, C5-H, J = 8.2 Hz), 12.20 (s, 1H, COOH, D2O-exchangeable). 13C NMR (DMSO-d6, 100 MHz, δ ppm): 117.02 (CH2'), 120.34 (C4), 123.40 (C6 + C7), 127.48, 127.70 (C3',5'), 129.01, 129.19 (C2',6'), 131.75 (C5,8), 133.31 (C1'), 134.77 (C8a), 135.00 (C4a), 141.59 (C2), 165.16 (C4=O), 170.50 (C=OOH). General Procedure for synthesis of 2-(2-phenyl-4-oxoquinazolin-3(4H)-yl)-N-Substituted acetamides 4a-f from 2-phenyl-4-oxo-3(4H)quinazolineacetyl chloride 2:A mixture of 2-phenyl-4-oxo-3(4H)quinazolineacetic acid 1 (1.40 g, 5 mmol) and thionyl chloride (3 mL) was heated under reflux for 1 hr. The deposited precipitate 2 was separated by filtration and used without further purification for the next step [1]. A solution of the acid chloride 2 (2.99 g, 10 mmol) in dry dioxane (10 mL) was treated with a solution of the appropriate amine (10 mmol) and triethylamine (1 mL) in dry dioxane (10 mL) and the mixture was heated for 1 h at 100oC, cooled and diluted with H2O. The obtained solid products were dried and crystallized from EtOH. IR (KBr, ύ, cm-1) for 4a-f: 3471-3329 (NH), 1774-1700, 1660-1643 (2 x C=O), 1636-1610 (C=N), 1604-1590, 1512-1497 (C=C Ar); in addition to: 1250, 1107 and 1090, 1049 (υas and υs C-O-C) for 4b,f; 3472-31210 (OH mixed with NH) for 4c,d.N-Phenyl 2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetamide 4a: Yield: 52% (1.85 g), M.p.: 230-232°C. IR (KBr, ύ, cm-1): 3341 (NH), 1660, 1654 (C=Os), 1597 (C=N), 1490 (C=C Ar). 1HNMR (DMSO-d6, 400 MHz, δ ppm): 7.11 (t, 1H, C7-H, J = 7.36 Hz), 7.15 (t, 1H, C6-H, J = 7.36 Hz), 7.29-7.94 (m, 12H, 10 Ar-Hs overlapping with CH2), 7.96 (d, 1H, C8-H, J = 7.96 Hz), 8.50 (d, 1H, C5-H, J = 8.20 Hz), 10.24 (s, 1H, NH, D2O-exchangeable). Anal. Calcd for C22H17N3O2: C, 74.35; H, 4.82; N, 11.82. Found: C, 74.53; H, 4.85; N, 12.01.N-(4-Methoxyphenyl)-2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetamide 4b: Yield: 63% (2.43 g), M.p.:159-161°C. IR (KBr, ύ, cm-1): 3329 (NH), 1762, 1643 (C=Os), 1600 (C=N), 1490 (C=C Ar). 1HNMR (DMSO-d6, 400 MHz, δ ppm): 3.75 (s, 3H, O-CH3), 6.93 (d, 2H, m- to OCH3, J = 9.08 Hz), 7.52 (t, 1H, C7-H, J = 7.52 Hz), 7. 56-7.75 (m, 8H, 5 Ar-Hs overlapping with C6-H + CH2), 7.69 (d, 2H, o- to OCH3, J = 9.0 Hz), 7.95 (d, 1H, C8-H, J = 7.08 Hz), 8.22 (d, 1H, C5-H, J = 7.08 Hz), 10.13 (s, 1H, NH, D2O-exchangeable). 13C NMR (DMSO-d6, 100 MHz, δ ppm): 55.68 (OCH3), 114.19 (CH2), 122.47 (C4'), 127.39, 127.43 (C3',5'), 128.00 (C3,5 of p-methoxyaniline), 128.29 (C7), 128.54 (C6), 128.80 (C2',6'), 129.40, 129.51 (C2,6 of p-methoxyaniline), 131.83 (C8), 132.68 (C5), 135.50 (C8a), 137.34 (C1'), 146.75 (C4a), 156.02 (C4 of p-methoxyaniline), 156.89 (C1 of p-methoxyaniline), 159.38 (C2), 165.59 and 167.63 (2 x C=O). Anal. Calcd for C23H19N3O3: C, 71.67; H, 4.97; N, 10.90. Found: C, 71.74; H, 5.28; N, 11.19.N-(2-Hydroxyethyl)-2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetamide 4c: Yield: 50% (1.62 g), M.p.: 185-187°C. IR (KBr, ύ, cm-1): 3352-3194 (NH+OH), 1650, 1635 (C=Os), 1590 (C=N), 1489 (C=C Ar). 1HNMR (DMSO-d6, 400 MHz, δ ppm): 3.39 (t, 2H, NH-CH2, J = 5.76 Hz), 3.55 (t, 2H, OH-CH2, J = 5.64 Hz), 4.78 (s, 2H, CH2), 7.21 (t, 1H, C7-H, J = 7.56 Hz), 7.60 (t, 1H, C6-H, J = 7.52 Hz), 7.52-7.96 (m, 6H, 5 Ar-Hs overlapping with C6-H), 7.88 (d, 1H, C8-H, J = 7.8 Hz), 8.67 (d, 1H, C5-H, J = 8.28 Hz), 8.86 (s, 1H, NH, D2O-exchangeable), 12.63 (s, 1H, OH, D2O-exchangeable). Anal. Calcd for C18H17N3O3: C, 66.86; H, 5.30; N, 13.00. Found: C, 67.11; H, 5.38; N, 13.23.N-(1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl)-2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetamide 4d: Yield: 58% (2.22 g), M.p.: 138-140°C. 1HNMR (DMSO-d6, 400 MHz,δ ppm): 4.45 (s, br, 6H, 3 x (OH-CH2) well resolved after D2O), 7.04 (t, 1H, C7-H, J = 7.32 Hz), 7.32 (t, 1H, C6-H, J = 7.44 Hz), 7.34-8.46 (m, 7H, 5 Ar-Hs overlapping with CH2), 7.88 (dd, 1H, C8-H, J = 7.8 Hz, 1.36 Hz), 8.07 (s, 1H, NH, D2O-exchangeable), 8.70 (d, 1H, C5-H, J = 8.12Hz), 14.83 (s, 1H, OH, D2O-exchangeable).Anal.Calcd for C20H21N3O5: C, 62.65; H, 5.52; N, 10.96. Found: C, 62.79; H, 5.58; N, 11.13.3-(2-Oxo-2-(piperazin-1-yl)ethyl)-2-phenylquinazolin-4(3H)-one 4e: Yield: 50% (1.74 g), M.p.: 278-280°C. 1HNMR (DMSO-d6, 400 MHz, δ ppm): 3.23 (s, 8H, 4 x CH2 well resolved into 2 singlets at 2.96 and 3.55 after D2O), 7.09 (t, 1H, C7-H, J = 7.32 Hz), 7.47 (t, 1H, C6-H, J = 6.96 Hz), 7.57 (s, 2H, CH2), 7.51-8.05 (m, 5H, 5 Ar-Hs overlapping with CH2), 8.18 (dd, 1H, C8-H, J = 8.8 Hz, 1.12 Hz), 8.73 (d, 1H, C5-H, J = 8.12 Hz), 14.24 (s, 1H, NH, D2O-exchangeable). 13C NMR (CDCl3, 100 MHz): 66.72 (4 x CH2 of piperazine), 119.25 (C4'), 122.94 (CH2), 123.07 (C6,7), 127.46 (C3',5'), 129.09 (C8), 129.36 (C2',6'), 131.84 (C5), 132.03 (C8a), 132.39 (C1'), 135.15 (C4a), 140.67 (C2), 165.20 (2 x C=O). Anal. Calcd for C20H20N4O2: C, 68.95; H, 5.79; N, 16.08. Found: C, 69.21; H, 5.85; N, 16.32.3-(2-Morpholino-2-oxoethyl)-2-phenylquinazolin-4(3H)-one 4f: Yield: 65% (2.27 g), M.p.: 176-178°C. 1H NMR (DMSO-d6, 400 MHz,δ ppm): 3.15 (t, 4H, 2 x CH2N, J = 4.92 Hz), 3.82 (t, 4H, 2 x CH2O, J = 5.0 Hz), 7.06 (ddd, 1H, C7-H, J = 7.68, 7.28, 1.08 Hz, becoming t after D2O at 7.07 ppm, J = 7.52 Hz, 7.48 Hz), 7.42 (ddd, 1H, C6-H, J = 1.68 and 7.32, 8.24 Hz, becoming t after D2O at 7.41 ppm, J = 7.84, 7.6 Hz), 7.57 (s, 2H, CH2 included within Ar multiplet), 7.52-7.62 (m, 3H, C3,4,5-Hs of 2-phenyl overlapping with CH2), 8.04 (dd, 2H, C2,6-Hs of 2- phenyl, J = 7.92 Hz, 1.76 Hz, becoming d after D2O at 7.98 ppm, J = 7.24 Hz), 8.11 (dd, 1H, C8-H, J = 7.76 Hz, 1.60 Hz, becoming d after D2O at 8.03 ppm, J = 7.72 Hz), 8.73 (dd, 1H, C5-H, J = 8.20 Hz, 0.72 Hz, becoming d after D2O at 8.63 ppm, J = 8.2 Hz). Anal. Calcd for C20H19N3O3: C, 68.75; H, 5.48; N, 12.03. Found: C, 69.01; H, 5.35; N, 12.32.2-(4-Oxo-2-phenylquinazolin-3(4H)-yl)acetohydrazide 5: Thionyl chloride (5 ml) was added to 2-phenyl-4-oxo-3(4H)-quinazolineacetic acid (1) (2.8 g, 10 mmol), and the solution was heated under reflux on a water bath for 1 h followed by removal of excess thionyl chloride under reduced pressure. Without further purification, the obtained acid chloride 2 was dissolved in dry pyridine (10 ml) and cooled to 0°C. To this solution, hydrazine hydrate (99%) (1.25 g, 1.23 ml, 25 mmol) was added at once with vigorous stirring. The mixture was stirred for 1 h and poured onto crushed ice with continuous stirring. The obtained product was filtered off, washed with H2O, dried and crystallized from EtOH; m.p. = 240-242°C; (reported m.p. = 242-245°C) [7], Yield 2 g (71%). IR (KBr, ύ, cm-1): 3317 (br, NH2, NH), 1652 (2 x C=O), 1603 (mixed vibration C=N + C=C Ar), 1529 (C=C Ar). 1H-NMR (CDCl3, 400 MHz, δ ppm): 3.12 (s, br, 2H, NH2, D2O-exchangeable), 7.08 (t, 1H, C7-H, J = 7.6 Hz), 7.28 (s, 2H, CH2), 7.48 (t, 1H, C6-H, J = 7.7 Hz, included within multiplet for 5 Ar-Hs), 7.52-7.60 (m, 5H, Ar-Hs), 8.05 (d, 1H, C8-H, J = 7.08 Hz), 8.79 (d, 1H, C5-H, J = 8.4 Hz), 11.86 (s, 1H, NH, D2O-exchangeable). EI-MS, m/z (% relative abundance): 294 [M+•] (1), 238 (11), 237 (61), 236 (58), 224 (16), 223 (19), 222 (16), 208 (29), 180 (18), 179 (19), 139 (11), 120 (16), 119 (54), 111 (16), 105 (100), 103 (18), 98 (10), 97 (17), 92 (14), 91 (12), 90 (18), 85 (13), 83 (18), 81 (12), 77 (97), 76 (38), 73 (19), 71 (23), 69 (25), 67 (14), 63 (16), 60 (21), 57 (40), 55 (37). Ethyl 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetyl)-hydrazinecarboxylate 6: Ethyl chloroformate (0.163g, 0.143 ml, 1.5 mmol) was added to a solution of acetohydrazide 5 (0.294 g, 1 mmol) and anhydrous K2CO3 (0.276 g, 2 mmol) in dry dioxane (5 mL) and the mixture was heated under reflux for 4 h. The reaction mixture was concentrated to half its volume, left to cool to R.T. and the separated product was filtered off and crystallized from dioxane; yield 60% (0.22 g), M.p.: 215-217oC. IR (KBr, ύ, cm-1): 3271 (NHs), 1715 (ester C=O), 1663 (2 x C=O), 1600 (mixed vibrational band for C=N + C=C Ar), 1444 (C=C Ar), 1251, 1041 (υas and υs C-O-C). 1H NMR (DMSO-d6, 300 MHz,δ ppm): 1.22 (t, dist, 3H, CH2-CH3), 4.11 (q, dist, 2H, CH2-CH3), 7.24 (t, 1H, C7-H, J = 7.5 Hz), 7.61 (t, 1H, C6-H, J = 7.0 Hz, overlapping with CH2 at 7.62), 7.56-7.82 (m, 5H, Ar-Hs), 7.93 (d, 1H, C8-H, J = 7.8 Hz), 8.66 (d, 1H, C5-H, J = 8.0 Hz), 10.66 (s, 1H, CONH, D2O-exchangeable), 12.00 (s, 1H, NHCOOEt, D2O-exchangeable). 13C NMR (DMSO-d6, 75 MHz):14.45 (CH3), 60.77 (COOCH2), 120.55 (CH2), 123.04 (C4 of 2- phenyl), 123.04 (C3,5 of 2- phenyl), 126.91 (C2,6 of 2- phenyl), 128.139 (C6,7), 128.94 (C5,8), 132.15 (C8a), 132.88 (C4a),134.36 (C1' of 2-phenyl), 139.34 (C2), 156.16 (C=O ester), 164.44 and 168.55 (2 x C=O). EI-MS m/z (relative abundance %):366 [M+•] (0.05), 236 (22), 224 (600, 223 (11), 208 (14), 180 (11), 179 (23), 146 (17), 119 (23), 111(18), 105 (100), 104 (13), 90 (17), 85 (19), 83 (17), 77 (92), 76 (29), 71 (29), 69 (21), 64 (18), 57 (47), 55 (22), 50 (18). Anal. Calcd for C19H18N4O4: C, 62.29; H, 4.95; N, 15.29; Found C, 62.35; H, 4.99; N, 15.41.2.2.4. Attempt preparation of N-substituted 2-(2-(4-oxo-2-phenyquinazolin-3(4H)-yl)acetylhydrazine-1-carboxamides 7a-e from ethyl 2-(2-(4-oxo-2-phenyl-quinazolin-3(4H)-yl)acetyl)hydrazine carboxylate 6: Glacial acetic acid (5 mL) was added slowly while shaking to a mixture of ethyl 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)acetyl)hydrazine carboxylate 6 (0.366 g, 1 mmol) and the appropriate 1° or 2° amine (1 mmol). The mixture was heated under reflux for 4-6 h, left to cool to R.T. and poured onto crushed ice. The obtained solid was filtered, dried and crystallized from EtOH. All products obtained had superimposable IR, 1H NMR and 13C NMR spectra. The structure was elucidated and found to be concordant with ethyl 2-(2-(4-oxo-2-phenylquinazolin-3(4H)-yl)-acetyl)hydrazine carboxylate 6. IR (KBr, ύ, cm-1): 3248 (NHs), 1744 (ester C=O), 1678 (2 x C=O), 1608 (C=N), 1596, 1474 (C=C Ar), 1258, 1064 (υas and υs C-O-C). 1H-NMR (DMSO-d6, 400 MHz) for products obtained from all reactions: 1.13-1.14 (t,3H, -CH3), 4.04-4.06 (q,dist, 2H, COOCH2), 7.51 (t, dist, 1H, C7-H, J = 7.5 Hz), 7.61-7.64 (t, 3H, C6-H, J = 7.0 Hz, overlapping with CH2), 7.49-7.99 (m, 5H, Ar-Hs), 7.79 (d, 1H, C8-H, J = 7.8 Hz), 8.20 (d, 1H, C5-H, J = 8.0 Hz), 10.05 (s, 1H, CONH, D2O-exchangeable), 10.47-10.48 (s, 1H, NHCOOEt, D2O-exchangeable).13C NMR (DMSO-d6, 100 MHz, δ ppm) for products obtained from reactions of the ester with tris-(hydroxymethyl)methyl amine and piperidine: 14.73 (CH3), 62.11 (COOCH2), 121.11 (CH2), 126.98 (C4 of 2- phenyl), 128.01 (C7), 128.21 (C6), 128.41 (C3,5), 129.00 (C2,6 of 2- phenyl), 129.10 (C8), 130.59 (C5), 134.03 (C4a,8a), 135.81 (C1 of 2- phenyl), 146.95 (C2), 155.92 (C=O ester), 156.90 and 160.27 (2 x C=O).Unexpectedly, treating ester 6 with aniline and glacial HOAc as previously mentioned above afforded 1-acetyl-4-phenyl semicarbazide 8 [8] instead of the amide 7a. Yield: 40% (0.77 g), M.p.: 168-170°C (Reported yield 30%); M.p.: 174.7-175.2°C. [8] IR (KBr, ύ, cm-1): 3294, 3260 (3 x NH), 1663, 1640 (2 x C=O), 1597, 1489 (C=C Ar). 1H NMR (DMSO-d6, 400 MHz) for compound 8: 2.04 (s, 3H, CH3), 7.02 (t, 1H, C4-H, J = 7.36 Hz), 7.28 (t, 2H, C3,5-Hs, J = 7.68 Hz),7.58 (d, 2H, C2,6-Hs, J = 7.90 Hz), 9.92 (s, 3H, NHs, D2O-exchangeable).13C NMR (DMSO-d6, 100 MHz) of 9: 24.45 (CH3), 119.44 (C3,5), 123.41 (C4), 129.10 (C2,6), 139.79 (C1), 168.72 (2 x C=O).
4. Conclusions
- In this study, the notably popular amidation reaction was explored for the preparation of some amides 4a-f, derived from quinazolinone starting from the ester 3 or the acid chloride 2. From this study, it appears that:- Amidation was first attempted, as previously reported, by reacting a mixture of ethyl (2-phenyl-4-oxoquinazolin-3(4H)-yl)acetate 3 and various 1° or 2° amines in hot refluxing glacial HOAc. The only product obtained in all trials was 4-oxo-2-phenyl-4(3H)-quinazolineacetic acid 1 instead of the required amides 4a-f assuming hydrolysis of the ester 3. This was confirmed by superimposition of all IR, 1H NMR and 13C NMR spectra of products obtained from all reactions (supplementary data). - To overcome the hydrolysis problem, amidation was carried out in alkaline medium and all proposed amides 4a-f were prepared in good yields upon treating acid chloride 2 with various 1° and 2° amines in presence of triethyl amine.Study also was expanded to determine the efficacy of the acid-catalyzed amidation reactions. Reacting ester 6 with the appropriate 1° or 2° amines in hot refluxing glacial HOAc did not afford the expected amides and recovery of the starting carbamate ester 6 was the only product obtained (supplementary data). In only one case, treating ester 6 with aniline in refluxing HOAc afforded 1-acetyl-4-phenylsemicarbazide 8 that had a melting point concordant with the one previously prepared.