-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2017; 7(1): 13-18
doi:10.5923/j.ajoc.20170701.03

New Triterpenoid from the Roots of Calotropis gigantea (L) Dryand (Asclepiadaceae)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLIman Omer1, 2, Ibrahim Abdurrahman1, 3, Yang Cai-Xia1
1College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou City, China
2School of Health Science, Ahfad Uuniversity for Women, Omdurman, Sudan
3Unit of Basic Science, Faculty of Agriculture, University of Zalingei, Zalingei, Sudan
Correspondence to: Yang Cai-Xia, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou City, China.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Seven triterpenoid derivatives were isolated from the roots of Calotropis gigantea (L) Dryand, a Chinese traditional medicinal plant. Their structures were established on the basis of their spectroscopic data and comparison with those in the literature. A new triterpenoid acetate derivative, compound 7, Calotropisis, was isolated and characterized by spectral methods.
Keywords: Calotropis gigantean, Acelepiadaceae, Triterpenoid derivatives
Cite this paper: Iman Omer, Ibrahim Abdurrahman, Yang Cai-Xia, New Triterpenoid from the Roots of Calotropis gigantea (L) Dryand (Asclepiadaceae), American Journal of Organic Chemistry, Vol. 7 No. 1, 2017, pp. 13-18. doi: 10.5923/j.ajoc.20170701.03.
Article Outline
1. Introduction
- Calotropis gigantea (L) Dryand (Asclepiadaceae) is mostly distributed in tropical and semitropical areas, mainly in Southwest China and South China in the wild [1-3]. It has been used in traditional medicine for the treatment of various ailments like parasitic diseases, digestive bloating from gas, cough, leprosy, and asthma by the people of the Li nationality, who are indigenous to Hainan island in China. Additionally, the plant is also used as a source of methane through anaerobic fermentation for bio fuel production [4]. This plant has been thoroughly explored for its many medicinal properties [5-7]. The chemistry of C. gigantea has been extensively investigated, leading to the isolation, of pregnanes [8, 9], flavonoids [10], a nonprotein amino acid [11], cardenolides [12-14] and terpenes [15-19]. In the present paper, we describe the isolation and structural elucidation of new triterpenoid acetates identified as calotropisis.
2. Experimental
2.1. Instrumentation and Materials
- NMR Spectra were recorded on a Bruker-DRX-400-NMR, (1H at 400Hz and 13C at 100Hz) spectrometer (Bruker Biospin Inc., Germany) and chemical shift values are given on a δ (ppm) scale with TMS as internal standard. 2D-NMR experiment was performed using standard Bruker micro-program (XWIN-NMR version 2.6 software. HR-EI-MS experiments were performed using a micro-mass–QTOF micro instrument, with an electro-spray ionization source (eV= 70 V, 80°C) (Waters Ltd., England). Column chromatography was carried out on silica gel (Merck kiesel gel 300-400 mesh, Qingdao Haiyang Chemical Group Company, China), TLCs were carried out on GF254 silica gel plates (Merck, Qingdao Haiyang Chemical Group Company, China). All solvents were of commercial grade and used after further purification by simple distillation.
2.2. Plant Material
- The Roots of C. gigantea were collected in August 2016 from Hunan province, south of China, and the plant was authenticated by prof. Chen Quan Yuan at the college of biology, Northwest Normal University, China, where a voucher specimen (No. 20141016) has been deposited in the herbarium of author’s laboratory.
2.3. Extraction and Isolation
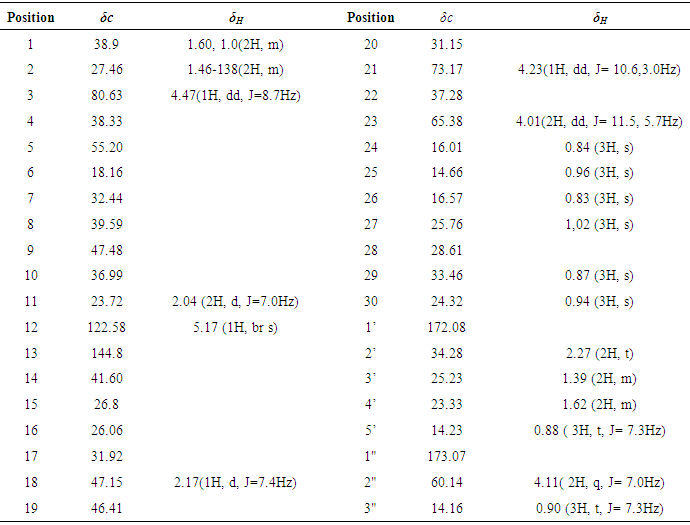
- The roots of the C. gigantean (Linn.) were air-dried for four weeks and ground into a powder. The root (15Kg) was sequentially extracted three times with 17 liters of ethanol at room temperature for 7 days each. Then, the extracts were filtered through cotton and concentrated with a rotary evaporator at 45C for removal of the organic solvent and dried. A total of 3kg of ethanolic extracts were dissolved in 5L hot distilled water and then prepared by successive partition with petroleum ether (PE) (40-60C) (fraction I), chloroform (fraction II), ethyl acetate (fraction III) and methanol (fraction IV). Each partition step was repeated three times to ensure complete extraction in each case. Fraction I was evaporated to yield 20g, which was chromatographed on 500g of silica gel (Merck kiesel gel 300-400 mesh) using (PE:C6H6) in a gradient elution (30:1-0:1) as the mobile phase. Three sub-fractions were obtained (A1, A2 and A3). Fraction A1 was again subjected to column chromatography, eluted with PE: EtOAC (gradient 30:1- 20:1) to obtained two further sub-fractions (A1-1–A1-2). Sub-fraction A1-1 chromatographed using a PE:EtOAc gradient (30:1-17:1) elution to yield compound 1. Sub-fraction A1-2 was chromatographed using a PE-EtOAc (17:1) elution and then recrystallized with a 1:1 mixture of MeOH:CHCl3 to obtain compound 2. Fraction A2 was also subjected to chromatography with a PE: EtOAc gradient (15:1-5-1) elution to yield compounds 3, 4 and 5 respectively. Fraction A3 was loaded onto a column and chromatography using a PE:EtOAC gradient (10:1-1:1) elution to provide compounds 6 and 7 respectively.Taraxasteryl acetate 1, obtained as white needle crystal, 34 mg, m.p 253-255°C. EI-MS m/z: 468 [M-CH3], C33H54O2. 1H-NMR: (400 MHz, CDCl3): δ 0.73 (3H, s, H-28), 0.79-083 (9H, s, H-23, 24, 29), 0.85 ( 3H, t , J= 7.3Hz, H-3'), 0.95 (3H, s, H-26), 0.97 (3H, s, H-25), 1,03 (3H, s, H-27), 2.21 (2H, t, H-2'), 4.05 (1H, dd, J= 8.7, 3.2 Hz, H-3), 4.53 (1H, br s, H-30). 13C-NMR (CDCl3, 100MHz) δ 38.34 (C-1), 23.31 (C-2), 80.61 (C-3), 37.65 (C-4), 55.29 (C-5), 18.18 (C-6), 33.60 (C-7), 40.0 (C-8), 50.03 (C-9), 37.57 (C-10), 21.33 (C-11), 25.8 (C-12), 38.84 (C-13), 41.95 (C-14), 26.58 (C-15), 39.14 (C-16), 34.41 (C-17), 48.59 (C-18), 38.25 (C-19), 154.4 (C-20), 25.65 (C-21), 39.35 (C-22), 27.88 (C-23), 16.60 (C-24), 15.38 (C-25), 16.25 (C-26), 14.06 (C-27), 26.08 (C-28), 19.43 (C-29), 107.23 (C-30), 171.92 (C-1'), 21.16 (C-2').α-amyrin acetate 2, obtained as white crystal, 154 mg, m.p 259-262°C. EI-MS m/z 468, C32H52O2. 1H-NMR: (400 MHz, CDCl3): δ 0.79 (3H, s, H-28), 0.86 (12H, s, H-23), 0.87(3H, s, H-24), 0.97(3H, s, H-29) ,1.00( 3H, s, H-30), 1.06 (3H, s, H-26) 1.24( 3H, s, H-25), 0.93 (3H, s, H-26), 0.96 (3H, s, H-25), 1,02 (3H, s, H-27), 2.03(3H, s, O-CH3), 2.27 (1H, dd, J = 7.6, 3.2 Hz, H-18), 4.50 (1H, dd, J= 8.7, 4.3 Hz, H-3), 5.12 (1H, br s, H-12). 13C-NMR (CDCl3, 100MHz) δ 38.45 (C-1), 28.73 (C-2), 80.33 (C-3), 38.45 (C-4), 55.24 (C-5), 18.23 (C-6), 32.85 (C-7), 40.01 (C-8), 47.41 (C-9), 36.80 (C-10), 23.36 (C-11), 124.25 (C-12), 139.49 (C-13), 42.5 (C-14), 26.91 (C-15), 26.85 (C-16), 33.7 (C-17), 59.04 (C-18), 39.59 (C-19), 39.62 (C-20), 31.23 (C-21), 41.5 (C-22), 28.05 (C-23), 15.63 (C-24), 15.47 (C-25), 16.8 (C-26), 23.5 (C-27), 28.38 (C-28), 17.04 (C-29), 21.3 (C-30), 170.94 (C-1'), 21.40 (C-2').α- amyrin caprylate 3, obtained as white crystal, 43 mg, m.p. 160-163°C. EI-Ms m/z 552, C38H64O2, 1H-NMR (400 MHz, CDCl3): δ 0.78 (3H, s, H-28), 0.85 (12H, s, H-23, 24, 29, 30), 0.88 (3H, t, J= 7.3Hz, H-8'), 0.94 (3H, s, H-26), 0.95 (3H, s, H-25), 1,15 (3H, s, H-27), 1.21-1.30 ( 10H, m, H-3'-7') 2.02 (2H, t, H-2'), 2.76 (1H, m, H-18), 4.49 (1H, dd, J= 8.7, 4.5 Hz, H-3), 5.11(1H, br s, H-12). 13C-NMR (CDCl3, 100MHz) δ 38.76 (C-1), 28.70 (C-2), 80.29 (C-3), 38.14 (C-4), 55.01 (C-5), 18.69 (C-6), 32.56 (C-7), 40.89 (C-8), 47.53 (C-9), 36.80 (C-10), 23.36 (C-11), 124.28 (C-12), 139.58 (C-13), 42.31 (C-14), 27.20 (C-15), 26.38 (C-16), 33.69 (C-17), 59.89 (C-18), 39.24 (C-19), 39.58 (C-20), 31.03 (C-21), 41.50 (C-22), 28.68 (C-23), 16.23 (C-24), 15.71 (C-25), 16.71 (C-26), 23.55 (C-27), 28.07 (C-28), 17.42 (C-29), 21.38 (C-30), 170.94 (C-1'), 34.42 (C-2'), 25.12 (C-3'), 29.24-29.33(C-4', 5', 6'), 23.69 (C-7'), 14.10 (C-8').11-oxo-α-amyrin ethylated 4, was obtained as white crystal, 31 mg, m.p. 278-281°C. EI-MS m/z 496, C33H52O3. 1H-NMR: (400 MHz, CDCl3): δ 0.79 (3H, s, H-28), 0.86 (12H, s, H-23, 24, 29, 30), 0.88 ( 3H, t , J= 7.3Hz, H-3') 0.94 (3H, s, H-26), 0.95 (3H, s, H-25), 1,15 (3H, s, H-27), 2.02 (3H, q, H-2'), 2.76 (1H, m, H-18), 4.50 (1H, dd, J= 10.7, 4.5 Hz, H-3), 5.57(1H, s, H-12). 13C-NMR (CDCl3, 100MHz) δ 38.93 (C-1), 28.68 (C-2), 80.89 (C-3), 38.24 (C-4), 55.23 (C-5), 18.92 (C-6), 32.35 (C-7), 40.21 (C-8), 47.20 (C-9), 36.92 (C-10), 199.55 (C-11), 130.36 (C-12), 164.85 (C-13), 42.01 (C-14), 27.11 (C-15), 26.43 (C-16), 33.33 (C-17), 59.03 (C-18), 39.60 (C-19), 39.18 (C-20), 31.22 (C-21), 41.67 (C-22), 28.80 (C-23), 16.25 (C-24), 15.53 (C-25), 16.90 (C-26), 23.67 (C-27), 28.37 (C-28), 17.45 (C-29), 21.27 (C-30), 170.90 (C-1'), 34. 36 (C-2'), 14.24 (C-3').1l, 12-Oxidotaraxerol acetate 5, obtained as colourless needles, 29 mg, m.p. over 300°C. EI-MS, m/z 482, C32H50O23. 1H-NMR (400MHz): 0.79, 0.84, 0.88, 0.90, 0 93, 0.98, 1.13, l.29 (each 3H,s, Me x 8), 2.02 (3H, s, OAc) 2.52(1H,d,5=4.4Hz, H-12), 2.98 (d, J = 4.7 Hz, 1H), 3.12 (lH, t, J= 44 Hz, H- 11), 4.50 (lH, dd, 5=10.5, 5.8Hz, H-3), 5.56 (lH, dd, 5=8.3, 3 5 Hz., H-15). 13C-NMR (CDCl3, 100MHz) δ 37.99 (C-1), 23.23 (C-2), 80.29 (C-3), 37.62 (C-4), 54.59 (C-5), 18.77 (C-6), 33.12 (C-7), 38.87 (C-8), 51.80 (C-9), 37.45 (C-10), 53.71 (C-11), 58.12 (C-12), 36.46 (C-13), 157.0 (C-14), 118.89 (C-15), 35.20 (C-16), 35.33 (C-17), 48.06 (C-18), 40.21 (C-19), 28.70 (C-20), 36.77 (C-21), 38.20 (C-22), 27.89 (C-23), 17.44 (C-24), 16.51 (C-25), 27.11 (C-26), 30.20 (C-27), 29.90 (C-28), 33.66 (C-29), 19.50 (C-30) 172.13 (C-1'), 20.96 (C-2').Germanicol acetate 6, was obtained as white powder, 36 mg, 279-283°C. EI-MS m/z 468, C32H52O2. 1H- NMR (400 MHz, CDCl3): δ 0.77 (3H,s, H-27), 0.84 (3H,s, H-24), 0.85 (3H,s, H-23), 0.92 (3H,s, H-26), 0.93 (3H,s, H-29), 0.97 (3H,s, H-30), 1.05 (3H,s, H-28), 1.09 (3H,s, H-25), 2.01 (3H, s, CH3-CO), 4.48 (1H, dd, J =11.5, 6.2 Hz, H-3), 5.35 (1H, s, H-19) . 13C-NMR (CDCl3, 100MHz) δ 38.29 (C-1), 23.40 (C-2), 80.24 (C-3), 37.52 (C-4), 55.25 (C-5), 18.29 (C-6), 33.60 (C-7), 40.31 (C-8), 50.20 (C-9), 37.04 (C-10), 21.35 (C-11), 26.38 (C-12), 39.60 (C-13), 42.20 (C-14), 26.71 (C-15), 38.16 (C-16), 34.34 (C-17), 48.05 (C-18), 39.25 (C-19), 156.86 (C-20), 25.73 (C-21), 38.84 (C-22), 27.07 (C-23), 16.62 (C-24), 16.32 (C-25), 15.89 (C-26), 14.59 (C-27), 18.90 (C-28), 25.73 (C-29), 109.57 (C-30), 170.73 (C-1'), 21.19 (C-2').Compound 7, obtained as a colorless powder, 33 mg, m.p. over 300°C, HR-ESI-MS (positive mode) m/z= 618.5548[M+NH4]+, 1H-NMR and 13C-NMR Table 1.
|
3. Results and Discussion
- The seven isolated compounds were identified as taraxasteryl acetate (1), α-amyrin acetate (2), α- amyrin caprylate (3), 11-oxo-α-amyrin ethylate (4), 1 l, 12-Oxidotaraxerol acetate (5), germanicol acetate (6), and Calotropisis (7). Figure 1 shows the structures of the triterpenoid isolated.
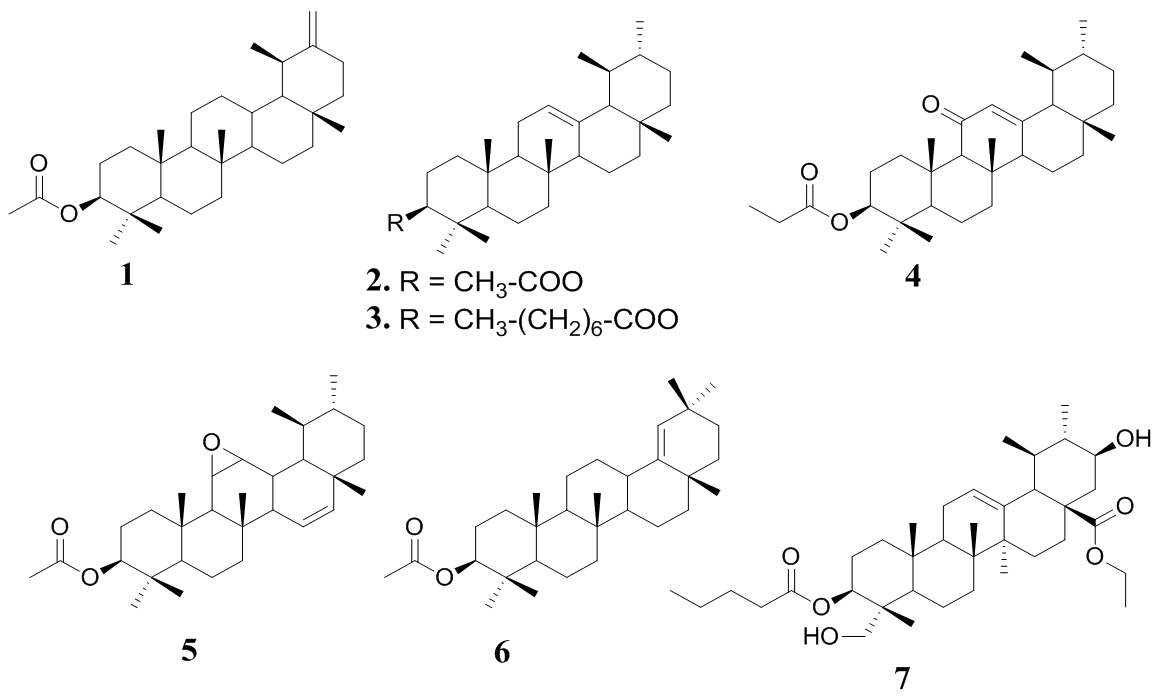 | Figure 1. Structures of compounds 1–7 |
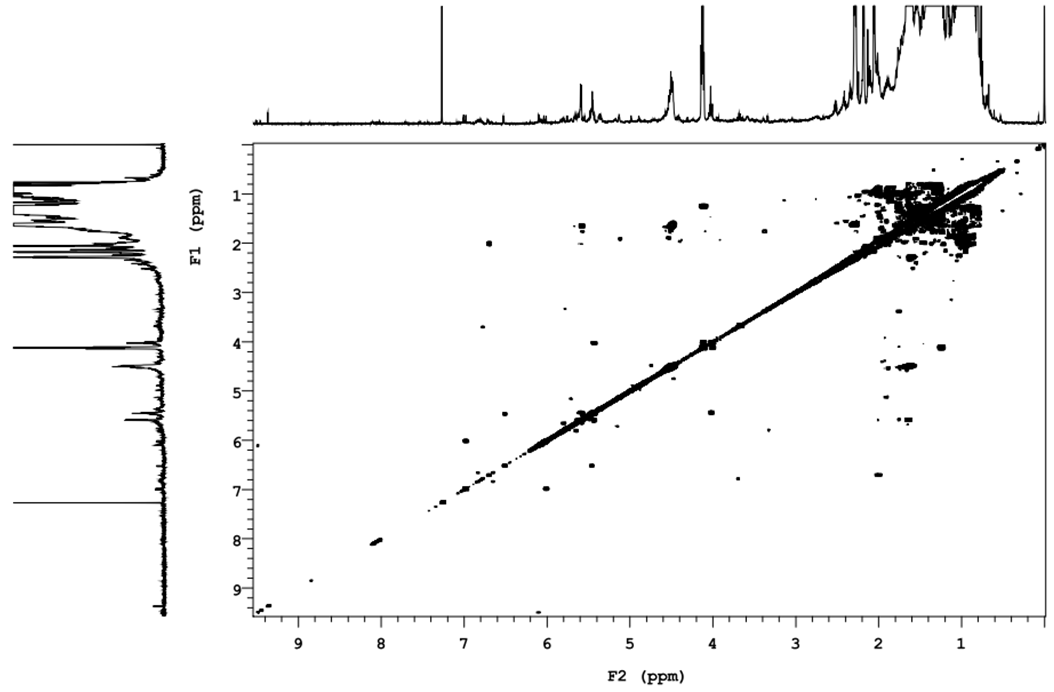 | Figure 2. 1H-1H COSY spectra of compound 7 |
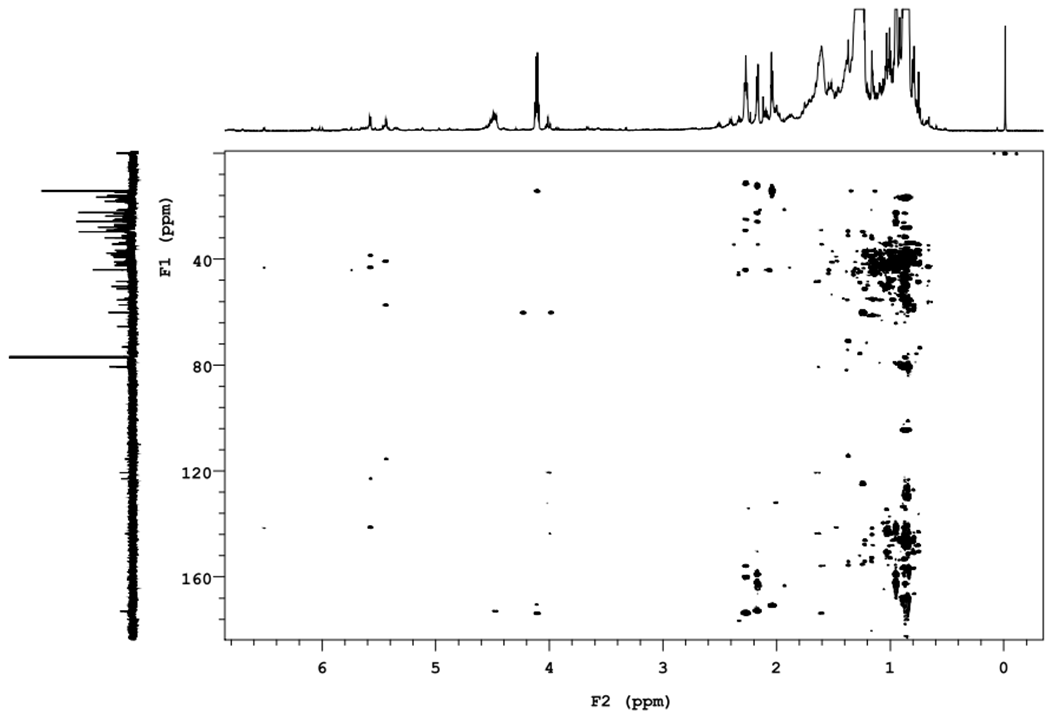 | Figure 3. HMBC spectra of compound 7 |
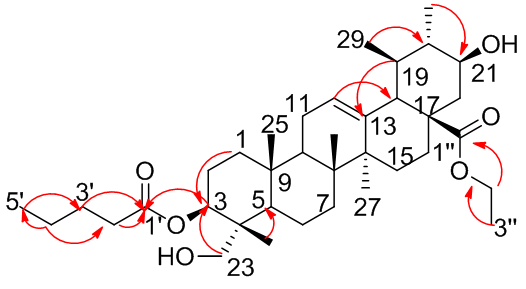 | Figure 4. The key HMBC correlations observed in compound 7 |
4. Conclusions
- Based on the results of our present study, roots of C. gigantean contain triterpenoid derivatives, as the principal secondary metabolites. A new triterpenoid acetate derivative, calotropisis with six known compounds were separated and their structures identified by spectral analysis.
ACKNOWLEDGEMENTS
- The authors are grateful to the China scholarship council (CSC) for the research scholarship. Authors are also grateful to College of Chemistry and Chemical Engineering, Northwest Normal University for providing the necessary facilities and infrastructure for the study.