-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2017; 7(1): 1-6
doi:10.5923/j.ajoc.20170701.01

Green Synthesis of a Cantharidine Model Compound Based on the Diels-Alder Reaction of Anti-Aromatic Tetracyclone and Maleimides as a Pharmacological Probe for Topical Application
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLUrbain C. Kasséhin1, 2, Hope A. Amou1, Fernand A. Gbaguidi1, Jacques H. Poupaert1, 2
1Laboratoire de Chimie Pharmaceutique Organique (MOCL), Ecole de Pharmacie, Faculté des Sciences de la Santé, Université d'Abomey-Calavi, Cotonou, Bénin
2Medicinal Chemistry Research Group (CMFA), Louvain Drug Research Institute (LDRI), Université Catholique de Louvain., Bruxelles, Belgium
Correspondence to: Urbain C. Kasséhin, Laboratoire de Chimie Pharmaceutique Organique (MOCL), Ecole de Pharmacie, Faculté des Sciences de la Santé, Université d'Abomey-Calavi, Cotonou, Bénin.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Using tetracyclone (2,3,4,5-tetraphenyl-2,4-cyclopentadien-1-one), and N-phenylmaleimide in a Diels-Alder (DA) approach, the synthesis of a lipophilic cantharidine-like phamacological probe has been designed to permit its topical use for the treatment of colon cancer or the West-African vector-borne parasitic trypanosomiasis. For the development of the target compound, special emphasis was put on implementation of green chemistry practices. This work underscored the anti-aromatic nature of tetracyclone and its consequences regarding the reactivity of this diene. Tuning of this peculiar DA reaction (n-butanol, 120°C, and 1.5 h) allowed to obtain selectively the endo-adduct with a final yield of 88%.
Keywords: Tetracyclone, N-phenylmaleimide, Cantharidine, Diels-Alder reaction, Anti-aromatic
Cite this paper: Urbain C. Kasséhin, Hope A. Amou, Fernand A. Gbaguidi, Jacques H. Poupaert, Green Synthesis of a Cantharidine Model Compound Based on the Diels-Alder Reaction of Anti-Aromatic Tetracyclone and Maleimides as a Pharmacological Probe for Topical Application, American Journal of Organic Chemistry, Vol. 7 No. 1, 2017, pp. 1-6. doi: 10.5923/j.ajoc.20170701.01.
Article Outline
1. Introduction
- In the course of a medicinal chemistry-driven program, our research consortium recently became interested in cyclopentadienone derivatives as a pivotal intermediate, which shares to some extent structural similarities with tetrcyclone (i.e. 2,3,4,5-tetraphenyl-2,4-cyclopentadien-1-one, 1). Tetraphenylcyclopentadienone (i.e. tetracyclone) is a dark purple to black crystalline solid that is poorly soluble in most organic solvents. The central cyclopentadienone core of the molecule is planar and conjugated with the four flanking phenyl groups. The C2-C3 and C4-C5 bond lengths are in the order of 1.35 Å, while the C1-C2, C3-C4, C5-C1 are closer to normal C-C single bonds with distances are around 1.5 Å [1]. The four flanking phenyl groups of 2,3,4,5-tetraphenyl-2,4-cyclopentadien-1-one adopt a propeller-like shape in their three-dimensional conformational behavior. The four peripheral phenyl rings are indeed rotated out of the plane of the central cyclopentadienone ring in a so-called gear-rotation situation because of the steric effect expressed by colliding ortho-hydrogen atoms (1,6-interaction, Pifzer strain) [2].In this paper, using tetracyclone as pivotal template and a Diels-Alder (DA) approach, we report our works concerned with the synthesis of a lipophilic cantharidine-like phamacological probe intentionally designed to be used topically (for example, by direct application within the intestinal lumen for the treatment of colon cancer) and more generally under the form of subcutaneously injected depot for the treatment of the Western African variant of the trypanosomiasis, which is a life-threatening parasitic (Trypanosoma brucei) vector-borne (tse-tse fly) disease and is responsible for the sleeping sickness in humans [3]. Throughout this contribution, special emphasis was put on implementation of "green chemistry" in our good laboratory practices (GLP) for the development of the target compound.
2. Results and Discussion
- This peculiar structure particularly drew our attention because of its anti-aromatic character in its dienic mesomeric form (Cfr left structure in Figure 1). Indeed, so far, the unsubstituted cyclopentadienone has remained a rather elusive structure due to its intrinsic anti-aromatic instability [4]. In support of the anti-aromatic, we can cite some spectroscopic evidence, and specially the UV-VIS spectrum which allows to evidence the existence of an internal charge-transfer. A charge-transfer complex or electron-donor-acceptor complex is an association of two molecules, or of different parts of the same molecule, in which a fraction of electronic charge is transferred externally or internally between the molecular entities. Many such complexes can undergo an electronic transition into an excited electronic state. The excitation energy of this transition occurs very frequently in the visible region of the UV-VIS spectrum, which produces the characteristic intense color and often deep fluorescence of such complexes. These optical absorption bands are often referred to as charge-transfer bands. UV-VIS spectroscopy is a powerful technique to characterize charge-transfer bands. In support of above assertion comes Figure 2: indeed, inspection of the Electrostatic Potential (ESP) contour mapping of 9-fluorenoneone shows that the carbonyl within the central cyclopentadienone motif is not conjugated with the internal dienic system.
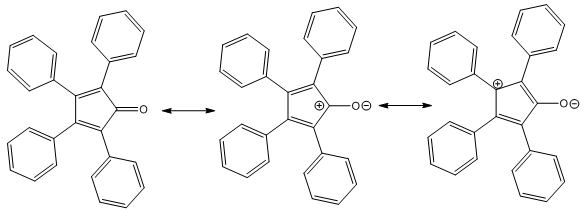 | Figure 1. A few representative canonical extreme mesomeric structures of tetracyclone generated according to the resonance theory |
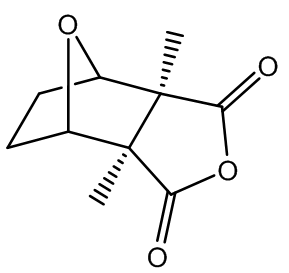 | Figure 2. Structure of cantharidine |
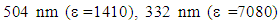 and
and 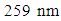
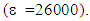 The highest wavelength
The highest wavelength 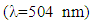 of low extinction commonly referred to as a “charge transfer band” can be assigned to the forbidden transition from the valence bonding to that of the conduction (polarized) bonding. The appearance of such an anomalous band is frequently considered as diagnostic predictor of an anti-aromatic contributing resonance structure and it is that absorbance which gives rise to the deep dark color of tetracyclone. The 332 nm
of low extinction commonly referred to as a “charge transfer band” can be assigned to the forbidden transition from the valence bonding to that of the conduction (polarized) bonding. The appearance of such an anomalous band is frequently considered as diagnostic predictor of an anti-aromatic contributing resonance structure and it is that absorbance which gives rise to the deep dark color of tetracyclone. The 332 nm 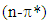 and 259 nm
and 259 nm 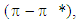 although showing a significant bathochromoic red shift, should not spark great concern at this level of our reasoning. There remains however a crucial question: if tetracyclone is in effect anti-aromatic, why does it exist and is so stable compared to unsubstituted cyclopentadienone? Reasons for such a stability are to be found firstly by the benzylic cationic character of the carbons at positions 2,3,4, and 5 owing to these four flanking phenyl groups, and secondly in the fact that tetracyclone is very insoluble and crystallizes out very quickly (even at high temperatures) from the reaction medium. Scavenged in the crystalline matrix, this high melting point compound (low motional capability in the solid matrix) does not dimerize and can be stored at room temperature for years without decomposition.As to the special anomalous nature of this “ketonic” carbonyl, we would like to point out its total lack of reactivity towards nucleophiles, even those with exceptional nucleophilicity and in particular those benefiting of the so-called “alpha-effect” such as 4-phenylthiosemicarbazide (4-PTSC). Indeed even with efficient catalysis (DMF.I2, anilinium chloride, anthranilic or sulfanilic acid, etc.) recently developed by our group or under forced harsh conditions (as for instance, refluxing in glacial acetic acid for 24h), tetracyclone opposed to 4-phenylthiosemicarbazone (4-PTSC) remained absolutely intact as judged by TLC, NMR and absence of the deep color discharge. Let us keep in mind that in the canonical extreme resonance structures, when the positive charge is located on a benzylic carbon (each four double bond carbons are eligible), this leaves the negatively charged oxygen in a stable enolate configuration and explains its non-reactivity. This total lack of reactivity towards 4-PTSC is quite reminiscent of the same puzzling behavior of the emblematic cyclopropylphenylketone, which suggests a potential participation (possibly through an anchimeric effect transmitted by a field effect) offered by the putative “sigma anti-aromaticity” of the cyclopropane ring. Indeed, the 1,3-dienic character of 1 has been largely exploited in the generation of various DA adducts. For example, 1 was used as a diene in the DA synthesis of 1, 2, 3, 4-tetraphenylnaphthalene with benzyne as dienophile [5] and it was equally employed to synthesize per-phenylbenzene (hexaphenylbenzene) using 1,2-diphenylacetylene [6]. Interestingly enough, in a similar way, pentaphenylpyridine derivatives were prepared via an unprecedented DA reaction between 1 and benzonitrile. Because of the high versatility of this reaction and the large chemical space potentially available via this route (i.e the chemodiversity centered on this template [7]), we successfully investigated a more flexible and "greener" access route to tetracyclone analogue derivatives in a previous publication [3].As to the Green Chemistry character of our present contribution, we would like to point out several avantages of our approach compared to previous contributions: (1) All solvents mentioned for running the key reaction are biodegradable and for the alcohols can be obtained by fermentation;(2) No solvent-costly chromatography was necessary for purifying the final product;(3) A rational plan was first designed before running experiments.In this connection, let us keep in mind that in principle the Diels–Alder reaction allows for generating in a single operation both the endo and exo diastereoisomeric species, a great asset in view of the ready availability of the automated separation technologies. The synthesis of tetracyclone (1) is more than well documented in the literature [8]. The title compound can be indeed prepared in principle even in good yield by consecutive aldol condensations be tween benzil and dibenzylketone (1,3-diphenyl-2-propanone) using a plethora of solvents and bases as homogeneous catalysts. Benzil is a standard building block in organic synthesis. A classic organic reaction of benzil is the benzilic acid rearrangement, in which base catalyses the conversion of benzil to benzilic acid [9]. This reactivity is exploited in the preparation of the drug phenytoin [10-11]. Benzil also reacts with 1, 3-diphenylacetone in an aldol condensation to give tetracyclone [12]. In our previous contribution, we provided the synthetic organic chemist community with a straightforward green method which we have taken advantage of in this note to study the potential accessibility of cantharidine mimeticks in an effort to generate a comprehensive compound library [13].The DA reaction remains in the realm of Synthetic Organic Chemistry a major player in creating new-formed carbon-carbon bonds. This remarkable reaction in its simplicity is the protypical example of a [4+2]
although showing a significant bathochromoic red shift, should not spark great concern at this level of our reasoning. There remains however a crucial question: if tetracyclone is in effect anti-aromatic, why does it exist and is so stable compared to unsubstituted cyclopentadienone? Reasons for such a stability are to be found firstly by the benzylic cationic character of the carbons at positions 2,3,4, and 5 owing to these four flanking phenyl groups, and secondly in the fact that tetracyclone is very insoluble and crystallizes out very quickly (even at high temperatures) from the reaction medium. Scavenged in the crystalline matrix, this high melting point compound (low motional capability in the solid matrix) does not dimerize and can be stored at room temperature for years without decomposition.As to the special anomalous nature of this “ketonic” carbonyl, we would like to point out its total lack of reactivity towards nucleophiles, even those with exceptional nucleophilicity and in particular those benefiting of the so-called “alpha-effect” such as 4-phenylthiosemicarbazide (4-PTSC). Indeed even with efficient catalysis (DMF.I2, anilinium chloride, anthranilic or sulfanilic acid, etc.) recently developed by our group or under forced harsh conditions (as for instance, refluxing in glacial acetic acid for 24h), tetracyclone opposed to 4-phenylthiosemicarbazone (4-PTSC) remained absolutely intact as judged by TLC, NMR and absence of the deep color discharge. Let us keep in mind that in the canonical extreme resonance structures, when the positive charge is located on a benzylic carbon (each four double bond carbons are eligible), this leaves the negatively charged oxygen in a stable enolate configuration and explains its non-reactivity. This total lack of reactivity towards 4-PTSC is quite reminiscent of the same puzzling behavior of the emblematic cyclopropylphenylketone, which suggests a potential participation (possibly through an anchimeric effect transmitted by a field effect) offered by the putative “sigma anti-aromaticity” of the cyclopropane ring. Indeed, the 1,3-dienic character of 1 has been largely exploited in the generation of various DA adducts. For example, 1 was used as a diene in the DA synthesis of 1, 2, 3, 4-tetraphenylnaphthalene with benzyne as dienophile [5] and it was equally employed to synthesize per-phenylbenzene (hexaphenylbenzene) using 1,2-diphenylacetylene [6]. Interestingly enough, in a similar way, pentaphenylpyridine derivatives were prepared via an unprecedented DA reaction between 1 and benzonitrile. Because of the high versatility of this reaction and the large chemical space potentially available via this route (i.e the chemodiversity centered on this template [7]), we successfully investigated a more flexible and "greener" access route to tetracyclone analogue derivatives in a previous publication [3].As to the Green Chemistry character of our present contribution, we would like to point out several avantages of our approach compared to previous contributions: (1) All solvents mentioned for running the key reaction are biodegradable and for the alcohols can be obtained by fermentation;(2) No solvent-costly chromatography was necessary for purifying the final product;(3) A rational plan was first designed before running experiments.In this connection, let us keep in mind that in principle the Diels–Alder reaction allows for generating in a single operation both the endo and exo diastereoisomeric species, a great asset in view of the ready availability of the automated separation technologies. The synthesis of tetracyclone (1) is more than well documented in the literature [8]. The title compound can be indeed prepared in principle even in good yield by consecutive aldol condensations be tween benzil and dibenzylketone (1,3-diphenyl-2-propanone) using a plethora of solvents and bases as homogeneous catalysts. Benzil is a standard building block in organic synthesis. A classic organic reaction of benzil is the benzilic acid rearrangement, in which base catalyses the conversion of benzil to benzilic acid [9]. This reactivity is exploited in the preparation of the drug phenytoin [10-11]. Benzil also reacts with 1, 3-diphenylacetone in an aldol condensation to give tetracyclone [12]. In our previous contribution, we provided the synthetic organic chemist community with a straightforward green method which we have taken advantage of in this note to study the potential accessibility of cantharidine mimeticks in an effort to generate a comprehensive compound library [13].The DA reaction remains in the realm of Synthetic Organic Chemistry a major player in creating new-formed carbon-carbon bonds. This remarkable reaction in its simplicity is the protypical example of a [4+2]  between a conjugated diene and a diversely substituted alkene, commonly termed the dienophile, to form a functionalized cyclohexene ring. This reaction was first disclosed by Otto Diels and Kurt Alder in 1928, for which they were awarded the Nobel Prize some 22 years later. This reaction now remains a “grand classic” among the concerted pericyclic reactions and it is by now commonly accepted that this reaction takes place generally via a single allegedly aromatic-like transition state, consequently without any intervention of one or several intermediates generated over the reaction coordinate of this transformation. As such, the DA reaction is governed by the law of orbital symmetry conservation and has been classified as a
between a conjugated diene and a diversely substituted alkene, commonly termed the dienophile, to form a functionalized cyclohexene ring. This reaction was first disclosed by Otto Diels and Kurt Alder in 1928, for which they were awarded the Nobel Prize some 22 years later. This reaction now remains a “grand classic” among the concerted pericyclic reactions and it is by now commonly accepted that this reaction takes place generally via a single allegedly aromatic-like transition state, consequently without any intervention of one or several intermediates generated over the reaction coordinate of this transformation. As such, the DA reaction is governed by the law of orbital symmetry conservation and has been classified as a 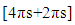 cycloaddition, for it proceeds via a suprafacial-suprafacial interaction of a
cycloaddition, for it proceeds via a suprafacial-suprafacial interaction of a 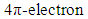 system with another
system with another 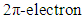 system, an interaction which is thermally allowed as a
system, an interaction which is thermally allowed as a 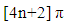 cycloaddition [14].The extent of sigma-pi delocalization along with the bond making-breaking process at the transition state are strongly influenced by the concomitant aromatization process going on. The aromaticity trends in the resulting transition state parallel the activation reaction energies; the extent of aromatization thus increases with increasing reaction rate and exothermicity. This implies that aromaticity (lowering the level of energy of the transition state) is the driving force governing most DA-cycloadditions. However, the title reaction involving an anti-aromatic substrate stabilized by the four flanking phenyl groups requires rather high temperatures (typically 120°C and over to proceed favorably in good yields). Moreover, when considering the reactive electrons involved in our peculiar reaction, there is a move from a predominance of the
cycloaddition [14].The extent of sigma-pi delocalization along with the bond making-breaking process at the transition state are strongly influenced by the concomitant aromatization process going on. The aromaticity trends in the resulting transition state parallel the activation reaction energies; the extent of aromatization thus increases with increasing reaction rate and exothermicity. This implies that aromaticity (lowering the level of energy of the transition state) is the driving force governing most DA-cycloadditions. However, the title reaction involving an anti-aromatic substrate stabilized by the four flanking phenyl groups requires rather high temperatures (typically 120°C and over to proceed favorably in good yields). Moreover, when considering the reactive electrons involved in our peculiar reaction, there is a move from a predominance of the  (in the reactants) to a
(in the reactants) to a  (in the product(s)) and therefore the transition state herein can be ascribed as a state of partial sigma-aromaticity, a concept first offered by pioneering US chemist Paul von Raguey-Schleyer.
(in the product(s)) and therefore the transition state herein can be ascribed as a state of partial sigma-aromaticity, a concept first offered by pioneering US chemist Paul von Raguey-Schleyer.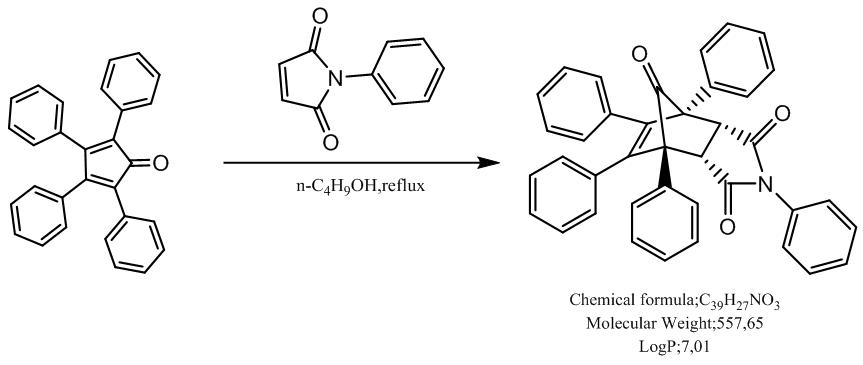 | Scheme 1. DA reaction of tetracyclone with N-phenylmaleimide (NPM) leading selectively to the endo-adduct |
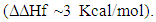 In this regard, a rather great similarity in the 13C-NMR spectrum of both species gave us confidence as to endo-configuration.
In this regard, a rather great similarity in the 13C-NMR spectrum of both species gave us confidence as to endo-configuration. | Figure 3. Electrostatic Potential Contour Mapping of 9-fluorenoneone showing that the carbonyl within the central cyclopentadienone motif is not conjugated with the internal double bonds |
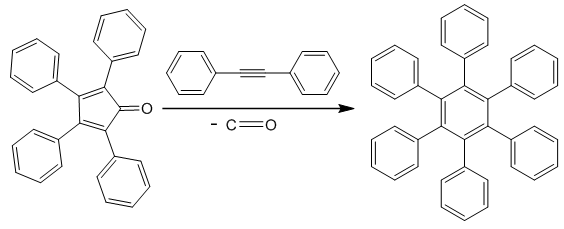 | Scheme 2. Synthesis of hexaphenylbenzene by DA reaction of tetracyclone with diphenylethylene |
3. Experimental Section
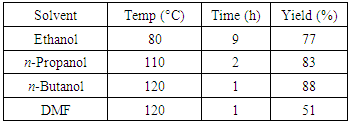
- Melting points were determined using an electrothermal melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer 457 spectrometer (London, UK) using vacuum-degassed anhydrous potassium bromide pellets. Wave numbers are expressed in cm−1. 1H- and 13C-NMR spectra were recorded at ambient temperature using a Brucker Spectrospin 400 spectrometer (Wissembourg, FR). Reported compounds were dissolved in CDCl3 or DMSO-d6 to obtain a 0.1 molar final solution. Chemical shifts are routinely expressed in the δ scale with TMS (tetramethylsilane) as internal standard. Thin layer chromatography (TLC) analyses were performed on Merck TLC plates (silica gel, 60 F 254, E. Merck, Darmstadt, DE, ref. 5735). For TLC, all compounds reported were insofar routinely monitored in two standard solvents, i.e. acetone/toluene/cyclohexane (solvent A, 5: 2: 3, v/v/v) and ethyl acetate/n-hexane (solvent B, 4: 6, v/v). The reverse-phase thin layer chromatography conditions were HPTLC plates RP-18 F-254 S (Merck) and methanol: water (75/25, v/v) as elution solvent. All compounds reported were found homogenous under such TLC and HPLC conditions. Specific reagents were all purchased from Aldrich (Milwaukee, WI, USA) or Acros (Geel, Belgium). All solvents respected ACS reagent grade specifications.Synthesis of TetracycloneInto a 250 ml round-bottomed flask equipped with a stirring bar, are placed 21.0g of benzil (0.1 mol), 23.1g of dibenzylketone (1,3-diphenyl-2-propanone, 0.11 mol) and 200 mL of absolute ethanol. Attach a water-cooled reflux condenser, and heat up the mixture in a hot water bath at 85°C until all solids get dissolved. Using a Pasteur pipette, add under rapid magnetic stirring and dropwise through the reflux condenser 25 mL of hot ethanolic potassium hydroxide (containing 5.7g of KOH, i.e. 0.12 mol) while keeping the water bath temperature still at 80°C, heat the solution for 15 min after which time, allow the mixture to cool down to room temperature, and then place it in an ice-water bath for 5 min. The solid product (a crystalline dark material) is isolated by vacuum filtration on a Büchner funnel, washed by small portions of ice-cold 95% ethanol, and dried overnight in a vacuum box at room temperature. Mp 219-220°C [20]. This material is chromatographically pure and its spectroscopic data are consistent with the proposed structure. 13C-NMR (CDCl3) 202.28 (C=0), 154.47 (C3, C4), 133.10 (C2, C5), 130.77 (arom. ipso carbon), 130.14 (arom. ipso carbon), 129.32, 128.47, 128.00, 127.44, 125.32 (arom. C-H carbons). UV-VIS in 0.02 M dioxane
 504 (1410), 332 (7080), 259 (26000).Reaction of tetracyclone with N-phenylmaleimide.(This procedure is representative of all similar operations shown in the Table 1.)
504 (1410), 332 (7080), 259 (26000).Reaction of tetracyclone with N-phenylmaleimide.(This procedure is representative of all similar operations shown in the Table 1.)
|
4. Conclusions
- In this paper, we explored the synthesis of a cantharidine-like model compound as a probe confined within the lumen of the intestine specifically designed to treat in a prophylactic manner the familial polyposis which induces colon and rectum cancer. This venture raised many interesting chemical problems, and in particular the anti-aromatic nature of tetracyclone, the substrate opposed to N-phenylmaleimide, and the underlying consequences. Initial positive results allows for the development of a concise compound library along this axis.
ACKNOWLEDGEMENTS
- The authors wish to acknowledge the generous contribution of the CTB/BTC (Coopération Technique Belge, Brussels, Belgium) both in terms of fellowship’s (U.C.K.) and financial support.