-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2016; 6(3): 93-101
doi:10.5923/j.ajoc.20160603.03

Synthesis of Halogenated 1H-Cyclohepta[2,1-b:3,4-b’]diindoles and Their Nucleophilic Aromatic Substitution Reactions
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLKosuke Ariyasu1, Ryuta Miyatake2, Yoshimitsu Kumai1, Akira Ohta1, Mitsunori Oda1
1Department of Chemistry, Faculty of Science, Shinshu University, Matsumoto, Japan
2Centre of Environmental Conservation and Research Safety, University of Toyama, Toyama, Japan
Correspondence to: Mitsunori Oda, Department of Chemistry, Faculty of Science, Shinshu University, Matsumoto, Japan.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Halogenated 1H-cyclohepta[2,1-b:3,4-b’]diindoles 5–8 were synthesized and their nucleophilic aromatic substitution reactions with amines and alcohols were studied. Compounds 5–7 were obtained by condensation of 2,2’-diindole (9) with 3-(dimethylamino)-2-haloacroleins in the presence of trifluoromethanesulfonic anhydride as a dehydrating reagent. On the other hand, 6,8-dichloro-1H-cyclohepta[2,1-b:3,4-b’]diindole (8) was synthesized in two steps from 9 viaenolketone 13. It was found that 8 underwent substitution reactions with amines and alcohols to afford the 6,8-substituted products in moderate to good yields, while compounds 5–7 showed reluctance to undergo substitution reactions. The phenomenon is rationalized by relative stability of the possible reaction intermediates based on DFT calculations.
Keywords: Biindole, Nucleophilic aromatic substitution, X-ray crystallographic analysis, Amine, DFT calculations
Cite this paper: Kosuke Ariyasu, Ryuta Miyatake, Yoshimitsu Kumai, Akira Ohta, Mitsunori Oda, Synthesis of Halogenated 1H-Cyclohepta[2,1-b:3,4-b’]diindoles and Their Nucleophilic Aromatic Substitution Reactions, American Journal of Organic Chemistry, Vol. 6 No. 3, 2016, pp. 93-101. doi: 10.5923/j.ajoc.20160603.03.
Article Outline
1. Introduction
- In 1999 Higa et al. reported isolation of a marine natural product, iheyamine A (1), from a colonial ascidian, Polycitorella sp. [1] The structure of 1 consists of two indole units around a unsaturated nitrogen-containing seven-membered ring in the middle of the molecule, having an aromatic 1,4-diazaazulene skeleton as a partial structure. In addition to its unique structural feature, 1 exhibits cytotoxic activity against tumor cells. [1] In respect to its prospective biological activity as an anti- cancer drug, various compounds (2–4) structurally related to 1 have been synthesized. (Figure 1) [2-4] We recently reported synthesis of non-substituted compound 4 and its spectroscopic properties. [4] In order to provide novel drug candidates having the structure of 4, we have continued our research on development of a divergent method for synthesizing its derivatives. Herein we describe the synthesis of halogen-substituted derivatives 5–8 and their nucleophilic aromatic substitution (SNAr) reactions. [5-6]
2. Results and Discussion
2.1. Synthesis of halogenated 1H-cyclohepta[2,1-b:3,4-b’] diindoles (5–8)
- Synthetic routes forming the central seven-membered ring to 7-halo- and 6,8-dichloro-1H-cyclohepta [2,1-b:3,4-b’] diindoles 5–8 from 2,2’-biindole (9) [7] are depicted in Schemes 1 and 2, respectively.Halogenated compounds, 5–7, were synthesized from 9 in a similar way of previously reported syntheses of 3 and 4. [4] Reactions of 9 with 3-(dimethylamino)-2-haloacroleins (10–12) in the presence of trifluoromethanesulfonic anhydride (Tf2O) in refluxing 1,2-dichloroethane (DCE) provided 5–7 in good yields. Also oxalyl chloride and phosphorus oxybromide could be used as a dehydrating reagent for syntheses of 5 and 6, respectively. However, when oxalyl chloride was used as a dehydrating reagent in the reaction of 9 and 11, a mixture of 5 and 6 formed probably by a halogen-exchanging reaction. Although Tf2O can be used in all annulation reactions, other dehydrating reagents should be selected for an appropriate substrate of 3-(dimethylamino)-2-haloacroleins. On the other hand, 6,8-dichloro compound 8 was synthesized in two steps from 9. Reaction of 9 with malonyl chloride in dichloromethane (DCM) produced 13 in 76% yield. The 1H-NMR spectrum of 13 indicates that 13 exists not as a diketone form but as a form of the keto-enol in solution, as depicted in Scheme 2. Subsequent reaction of 13 with phosphorus oxychloride in the presence of ethyldiisopropylamine provided 8 in 58% yield. Unfortunately, similar reaction of 13 with phosphorus oxybromide instead of phosphorus oxychloride did not give any clear product. Structures of all new halogenated compounds 5–8 were confirmed by spectroscopic analyses.
2.2. Nucleophilic Aromatic Substitution Reactions (SNAr) of 5–8
- Like halopyridines, halogenated 1-azaazulenes and 2,3-benzo-1-azaazulenes are known to behave as good acceptors against nucleophiles in their substitution reactions. [8-11] Therefore, we expected that the halogenated compounds 5–8 could also undergo SNAr reactions. Indeed, dichloride 8 reacts secondary amines to give diamino derivatives 14–18 in good to high yields (Scheme 3).
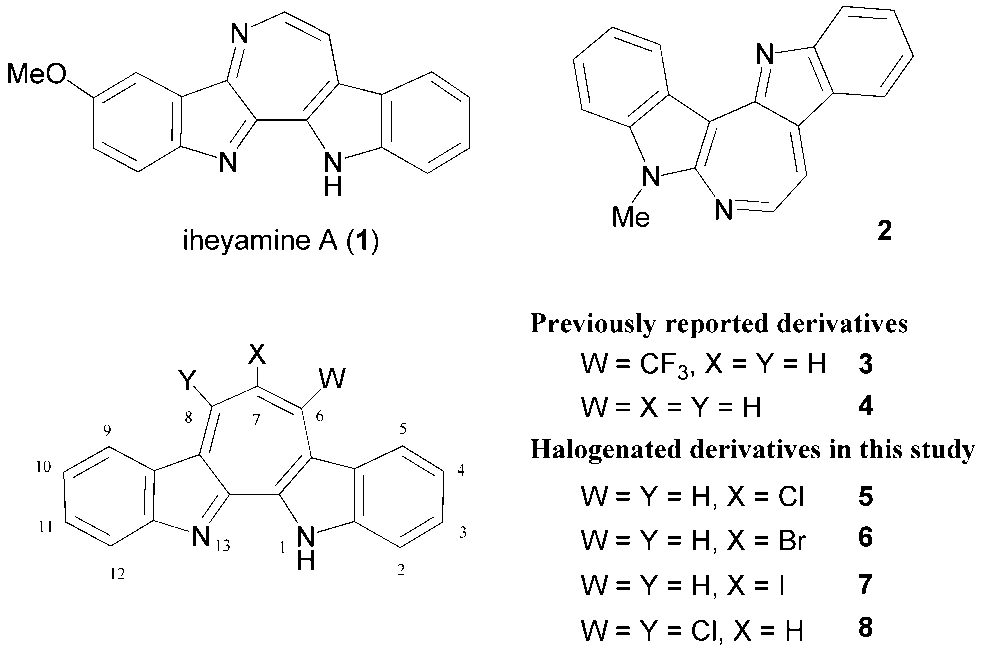 | Figure 1. Iheyamine A (1) and structurally related synthetic compounds (2–8) |
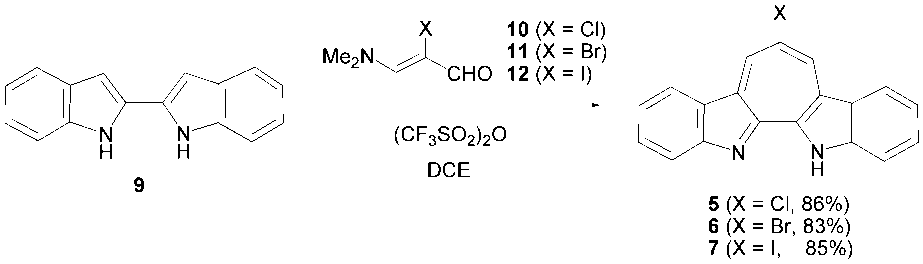 | Scheme 1. Synthesis of 5–7 |
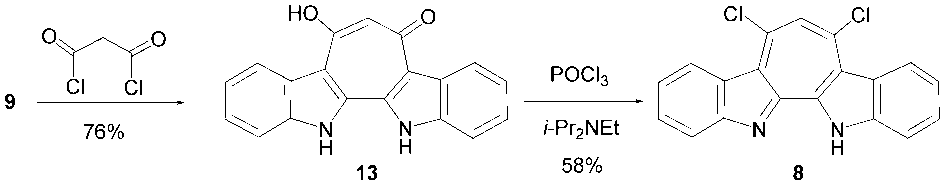 | Scheme 2. Synthesis of 8 |
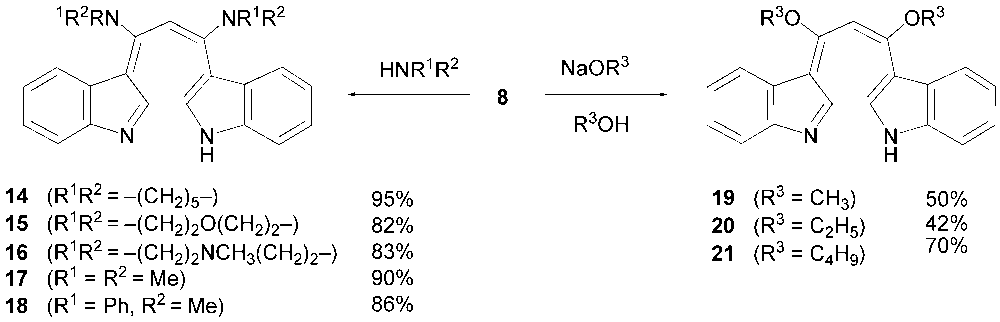 | Scheme 3. The nucleophilic substitution reactions of 8 |
 | Scheme 4. Synthesis of unsymmetrical derivatives |
|
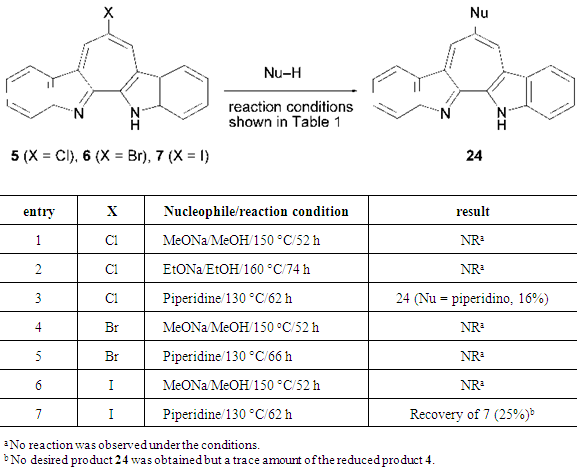
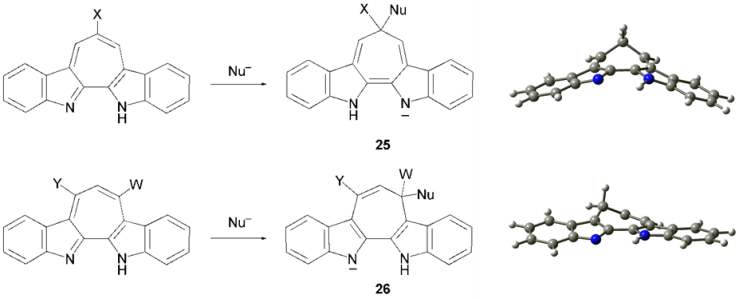 | Scheme 5. Expected anionic intermediates in the substitution reactions and the calculated structures of their simplified molecules (right) |
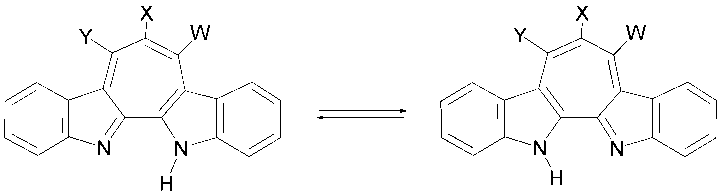 | Scheme 6. Tautomerism of 1H-cyclohepta[2,1-b:3,4-b’]diindoles |
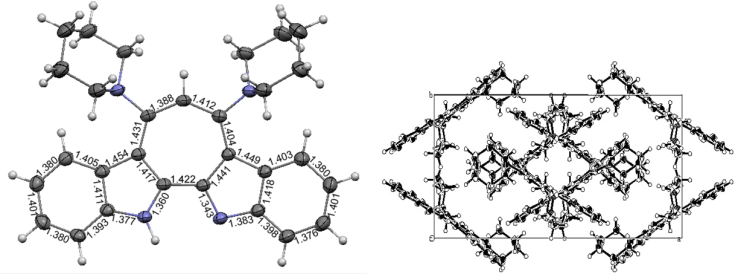 | Figure 2. ORTEP drawing of 14 with bond lengths (left) and its packing diagram viewed along c axis (right) |
3. Experiments
3.1. General
- Melting points were measured on a Yanaco MP-3 and are uncorrected. IR spectra were recorded on JEOL Diamond-20 and JASCO FT/IR-4100 spectrometers. UV-vis spectra were measured on a Shimadzu UV-2550 spectrometer. 1H- and 13C-NMR spectra were recorded on JEOL λ400 and ECA500 spectrometers. Chemical shift values of tetramethylsilane (δ = 0 ppm) for 1H-NMR spectra and CDCl3 (δ = 77.0 ppm) for 13C-NMR spectra were used as internal standard. Mass spectra were measured on a JMS-700 mass spectrometer. Column chromatography was performed with Silica gel 60N from Kanto Chem. DCM, DCE and DMF were purchased from Kanto Chem. and was distilled over CaH2. Dimethylaminoacrolein and oxalyl chloride were purchased from Wako Chemical Co. and were used without purification. Tf2O and N-halosuccinimides were purchased from Tokyo Chemical Industry, Inc. 2,2’-Biindole was prepared in two steps from 2-methylaniline according to a procedure reported by Bergman et al. [5]
3.2. 3-(Dimethylamino)-2-haloacroleins 10–12
- 3-(Dimethylamino)-2-haloacroleins 10–12 were prepared from 3-(dimethylamino)acrolein by halogenation with N-halosuccinimides.[17–19] Following are selected spectral data of 10–12.2-Chloro-3-(dimethylamino)acrolein 10: Colorless solids, mp = 45–48°C [lit. 40–43°C] [17]; 1H NMR (500 MHz, CDCl3) δ = 3.36 (br, 6H), 6.89 (s, 1H), 8.69 (s, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ = 39.3, 47.2, 104.2, 154.0, 183.0 ppm; MS (70 eV) m/z (%) = 135 (M+, 33), 133 (M+, 100), 118 (45), 98 (21), 89 (17), 55 (14). HRMS Calcd for C5H835ClNO 133.0294; found 133.0292.2-Bromo-3-(dimethylamino)acrolein 11: Colorless needless, mp = 65–68°C [lit. 54.5–56°C] [18]; 1H NMR (500 MHz, CDCl3) δ = 3.31 (br, 6H), 7.12 (s, 1H), 8.79 (s, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ = 40.0, 47.4, 92.9, 155.9, 183.4 ppm; MS (70 eV) m/z (%) = 179 (M+, 51), 177 (M+, 53), 162 (36), 160 (35), 98 (100), 55 (38). HRMS Calcd for C5H879BrNO 176.9789; found 176.9787.3-(Dimethylamino)-2-iodoacrolein 12: Colorless needless, mp = 74–76°C [19-20]; 1H NMR (500 MHz, CDCl3) δ = 3.34 (br, 6H), 7.34 (s, 1H), 8.29 (s, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ = 42.4, 46.7, 66.8, 159.4, 185.6 ppm; MS (70 eV) m/z (%) = 225 (M+, 49), 127 (7), 118 (45), 98 (100), 68 (8), 55 (22). HRMS Calcd for C5H8INO 224.9651; found 224.9649.
3.3. 7-Halo-1H-cyclohepta[2,1-b;1,2-b’]diindoles (5–7)
- A solution of 3-(dimethylamino)-2-haloacrolein (2.00 mmol) in 4 ml of DCE was added dropwise to a solution of Tf2O (336 μL, 2.00 mmol) in 3 ml of DCE at 0°C. After being stirred at room temperature for 15 min, a suspension of 9 (697 mg, 3.00 mmol) in 20 ml of DCE was added to the reaction solution. Then, the reaction mixture was heated to 100oC for 1 h under nitrogen atmosphere. After being cooled to room temperature, the reaction mixture was poured into a saturated sodium bicarbonate solution (30 ml) and extracted with chloroform (100 ml x 3). The combined organic layer was washed with brine and dried over Na2SO4. Then, the solvent was removed under reduced pressure and the residue was crystallized from chloroform to give the product as dark red microcrystals. The filtrate was purified by silica gel column chromatography. Elution with ethanol/chloroform (7/93) provided the product further. 5: Dark red microcrystals, mp = 279–283°C; 1H NMR (500 MHz, CDCl3) δ = 7.43–7.53 (m, 6H), 8.32 (d, J = 7.8 Hz, 2H), 8.97 (s, 2H) ppm [21]; 13C NMR (126 MHz, CDCl3) δ = 116.2, 120.3, 122.1, 126.9, 127.0, 129.0, 129.6, 130.8, 144.2, 146.9 ppm; IR (KBr) νmax = 1397vs, 1362vs, 1234s, 1194s, 741vs cm–1; UV-vis (CH3CN) λmax = 227 (logε = 4.51), 321 (4.72), 335 (4.69), 366 (4.23), 408 (3.58), 433 (3.52), 517 (3.67) nm; MS (70 eV) m/z (%) = 304 (M+, 34), 302 (M+, 100), 266 (16), 151 (10). HRMS Calcd for C19H1135ClN2 302.0611; found 302.0609.6: Dark red plates, mp = 284–288°C; 1H NMR (400 MHz, CDCl3) δ = 3.75 (br, 1H), 7.48 (t, J = 7.8 Hz, 2H), 7.59 (t, J = 7.8 Hz, 2H), 7.64 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 9.11 (s, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ = 115.6, 115.9, 120.4, 122.3, 126.6, 129.2, 130.0, 133.7, 144.4, 149.0 ppm; IR (KBr) νmax = 1398vs, 1363vs, 1192s, 745vs cm–1; UV-vis (MeCN) λmax = 284sh (logε = 4.22), 298sh (4.40), 322 (4.72), 337 (4.70), 366 (4.24), 408 (3.58), 434 (3.52), 518 (3.69) nm; MS (70 eV) m/z (%) = 348 (M+, 98), 346 (M+, 100), 266 (31), 240 (18), 173 (11), 133 (12). HRMS Calcd for C19H1179BrN2 346.0106; found 346.0107.7: Dark red microcrystals, mp = 284–287°C; 1H NMR (500 MHz, CDCl3) δ = 7.45–7.47 (m, 2H), 7.53-7.57 (m, 4H), 8.29 (d, J = 7.8 Hz, 2H), 9.27 (s, 2H) ppm [21]; 13C NMR (500 MHz, CDCl3) δ = 87.0, 116.1, 120.3, 122.2, 126.5, 128.9, 130.8, 139.8, 144.5, 146.7 ppm; IR (KBr) νmax = 1474s, 1403s, 1363s, 1241s, 1193s, 1107s, 755s, 745vs, cm–1; UV-vis (CH3CN) λmax= 223 (logε = 4.41), 251sh (4.07), 323 (4.66), 338 (4.62), 367 (4.18), 408sh (3.51), 435 (3.42), 519 (3.58) nm; MS (70 eV) m/z (%) = 394 (M+, 100), 267 (33), 260 (22), 240 (16). HRMS Calcd for C11H19IN2 393.9967; found 393.9967.
3.4. 1,6,13-Trihydro-8-hydroxycyclohepta[2,1-b;3,4-b’]- diindol-6-one (13)
- To a solution of malonyl chloride (617 mg, 4.40 mmol) in 20 ml of DCE was added a suspension of 9 (929 mg, 4.00 mmol, 1 eq.) in 15 ml of DCE with cooling on an ice bath. After refluxing of the reaction mixture under nitrogen atmosphere for 3.5 h, the solvent was removed under reduced pressure. The residue was homogenously suspended in 30 ml of a saturated sodium bicarbonate solution and solids were collected by filtration. The solid obtained was washed with water (20 ml), acetone (20 ml) and ethanol (20 ml), and dried under vacuum to give 907 mg (76%) of 13 as brown solids. Mp > 300°C (decomp.). 1H NMR (500 MHz, DMSO-d6, 60°C) δ = 3.75 (brs, 2H, N-H & O-H), 7.47 (t, J = 7.8 Hz, 2H), 7.51 (s, 1H), 7.65 (t, J = 7.8 Hz, 2H), 7.82 (d, J = 7.8 Hz, 2H), 8.66 (d, J = 7.8 Hz, 2H) ppm; 13C NMR (126 MHz, DMSO-d6, 60°C) δ = 105.4, 111.8, 114.9, 122.5, 124.40, 124.43, 127.3, 133.6, 137.3, 168.3 ppm; IR (KBr) νmax = 3158s, 3097s, 3076s, 3030s, 2983s, 1495s, 1455s, 1426s, 1389vs, 1359s, 1324s, 1248s, 1223s, 748s cm–1; UV-vis (EtOH) λmax = 213 (logε = 4.42), 230sh (4.27), 300 (4.76), 324sh (4.19), 339sh (4.05), 349sh (4.02), 367 (4.10), 385 (4.18) nm; MS (FAB) m/z (%) = 301 (MH+, 32), 153 (81), 136 (100), 107 (37), 73 (87), 51 (23). HRMS Calcd for C19H13N2O2+ 301.0972; found 301.0977.
3.5. 6,8-Dichloro-1H-cyclohepta[2,1-b;3,4-b']diindole (8)
- Phosphorus oxychloride (1.44 ml, 15.4 mmol) was carefully added to a mixture of i-Pr2EtN (0.54 ml, 3.1 mmol) and 13 (463 mg, 1.54 mmol) with cooling on an ice bath. The reaction mixture was heated at 100°C for 2 h under argon atmosphere. After being cooled to room temperature, the reaction mixture was poured into a 30 ml of saturated sodium carbonate aqueous solution and extracted with chloroform (40 ml x 3). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography. Elution with ethanol/chloroform (5:95) gave 301 mg (58%) of 8 as red prisms. Mp = 279–282°C. 1H NMR (500 MHz, CDCl3, 40°C) δ = 7.55 (ddm, J = 8.4, 7.2 Hz, 2H), 7.74 (ddm, J = 8.1, 7.2 Hz, 2H), 7.96 (dm, J = 8.1 Hz, 2H), 8.14 (s, 1H), 8.94 (dm, J = 8.4 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ = 114.8, 124.42, 124.46, 125.6, 126.7, 130.1, 131.3, 137.6, 142.1, 143.4 ppm; IR (KBr) νmax = 1559s, 1396vs, 1373vs, 1111s, 740vs cm–1; UV-vis (CH3CN) λmax = 225 (logε = 4.41), 261 (4.05), 322 (4.66), 342 (4.60), 370sh (4.15), 408 (3.65), 435 (3.60), 508 (3.64) nm; MS (70 eV) m/z (%) = 340 (M+, 12), 338 (M+, 65), 336 (M+, 100), 266 (26), 265 (12). HRMS Calcd for C19H1035Cl2N2 336.0221; found 336.0226.
3.6. 6,8-Dipiperidino- 6,8-dimoripholino-, and 6,8-bis (N-methylpiperzino)-1H-cyclohepta[2,1-b;3,4-b']-diindoles (14–16)
- A mixture of 8 (30 mg, 0.089 mmol) and piperidine (1.0 ml) was heated at 120°C for 1 h in a sealed tube purged by argon. After being cooled to room temperature, the reaction mixture was poured into 40 ml of 0.2 M hydrochloric acid. The resulted mixture was extracted with chloroform (20 ml x 3), and the combined organic layer was washed with a saturated sodium bicarbonate solution (20 ml x 2) and was dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by silica gel chromatography. Elution with ethanol/chloroform (5/95) gave 37 mg (95%) of 14 as orange prisms. Mp = 271–272°C (decomp.). 1H NMR (500 MHz, DMSO-d6, 100°C) δ = 1.72 (quin, J = 5.5 Hz, 4H), 1.87 (quin, J = 5.5 Hz, 8H), 3.38 (t, J = 5.5 Hz, 8H), 7.13 (s, 1H), 7.29 (t, J = 7.8 Hz, 2H), 7.43 (t, J = 7.8 Hz, 2H), 7.79 (d, J = 7.8 Hz, 2H), 8.47 (d, J = 7.8 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ = 24.5, 26.3, 54.4, 107.4, 115.3, 119.3, 120.7, 123.2, 125.8, 126.6, 144.7, 144.9, 159.3 ppm; IR (KBr) νmax = 1556s, 1532s, 1449s, 1409s, 1362s, 1341s, 1240s, 1225s, 1199s, 1122s, 1102s, 749s cm–1; UV-vis (CH3CN) λmax = 224 (logε = 4,49), 237sh (4.35), 333 (4.86), 357sh (4.48), 417 (4.04), 471sh (3.78) nm; MS (70 eV) m/z (%) = 434 (M+, 100), 433 (11), 365 (12), 351 (16), 294 (10), 281 (12),. 281 (12), 268 (17), 217 (9). HRMS Calcd for C29H30N4 434.2471, found 434.2476.Compounds 15–16 were synthesized similarly from 8 with morpholine and N-methylpiperzine in 82 and 90% yields, respectively. 15: Orange prisms, mp = 271–272°C (decomp.). 1H NMR (500 MHz, DMSO-d6, 100°C) δ = 3.39 (t, J = 4.4 Hz, 8H), 3.99 (t, J = 4.4 Hz, 8H), 7.08 (s, 1H), 7.34 (t, J = 7.6 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.81 (d, J = 7.6 Hz, 2H), 8.54 (d, J = 7.6 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3, 50°C) δ = 53.12, 67.0, 106.2, 115.6, 119.7, 121.3, 123.3, 126.2, 126.6, 145.0, 145.3, 158.1 ppm; IR (KBr) νmax = 1534s, 1445s, 1374s, 1345s, 1332s, 1220s, 1208s, 1029s, 887s, 770s, 746s cm–1; UV-vis (CH3CN) λmax = 225 (logε = 4.45), 331 (4.82), 357sh (4.45), 378sh (4.08), 395 (3.97), 418 (3.95), 474sh (3.72) nm; MS (70 eV) m/z (%) = 438 (M+, 100), 295 (10), 294 (11), 281 (10). HRMS Calcd for C27H26N4O2 438.2056, found 438.2052.16: Orange solids, mp = 245°C (decomp.). 1H NMR (500 MHz, CDCl3) δ = 2.47 (s, 6H), 2.77 (br, 4H), 2.86 (br, 4H), 3.30 (br, 4H), 3.64 (br, 4H), 7.17 (s, 1H), 7.35 (t, J = 7.5 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H) 7.57 (d, J = 7.5 Hz, 2H), 8.61 (d, J = 7.5 Hz, 2H) ppm. 13C NMR (126 MHz, CDCl3) δ = 46.2, 52.7, 55.2, 106.5, 115.5, 119.3, 120.9, 123.2, 126.1, 144.8, 145.0, 158.3 ppm. IR (KBr) νmax = 1449s, 1357s, 1201s, 1008s, 751s cm–1. UV-vis (CH3CN) λmax = 224 (logε = 4.47), 332 (4.83), 356sh (4.47), 417 (3.97), 464 (3.75) nm. MS (70 eV) m/z (%) = 464 (94, M+), 394 (100). HRMS Calcd for C29H32N6 464.2689; found 464.2691.
3.7. 6,8-Bis(dimethylamino)-1H-cyclohepta[2,1-b;3,4-b']- diindole (17)
- A mixture of 8 (20 mg, 0,059 mmol), dimethylamine hydrochloride (11 mg, 0.12 mmol) and triethylamine (24 mg, 0.24 mmol) in 2 ml of dry DMF was heated to 100°C for 2 h under nitrogen atmosphere. After being cooled to room temperature, the reaction mixture was poured into water (5 ml) and the precipitate formed was filtered off. The solid was purified by alumina column chromatography. Elution with methanol/dichloromethane (4/96) gave 19 mg (90%) of 17 as orange solids. Mp = 245–250°C. 1H NMR (500 MHz, CDCl3) δ = 3.18 (s, 6H), 6.80 (s, 1H), 7.31 (t, J = 7.7 Hz, 2H), 7.36 (t, J = 7.7 Hz, 2H), 7.60 (d, J = 7.7 Hz, 2H), 8.19 (d, J = 7.7 Hz, 2H) ppm [21]; 13C NMR (126 MHz, CDCl3) δ = 44.6, 104.5, 115.3, 116.7, 120.5, 122.8, 125.0, 126.1, 144.2, 144.5, 158.4 ppm; IR (KBr) νmax = 1556s, 1402s, 1354vs, 1120s, 750s cm–1; UV-vis (CH3CN) λmax = 226 (logε = 4.42), 236sh (4.35), 332 (4.82), 414 (3.96), 466 (3.71) nm; MS (70 eV) m/z (%) = 354 (M+, 100), 353 (13), 339 (19), 310 (9), 296 (16), 268 (11). HRMS Calcd for C23H22N4 354.1845; found 354.1841.
3.8. 6,8-Bis(N-methylanilino)-1H-cyclohepta-[2,1-b;3,4- b']diindole (18)
- A mixture of 8 (10 mg, 0,029 mmol) and N-methyaniline (1 mL) was heated to 120°C for 4 h under argon atmosphere. After being cooled to room temperature, the reaction mixture was poured into 1N HCl (5 ml) and was extracted with chloroform (20 mL x 3). The combined organic layer was washed with a saturated sodium bicarbonate solution (20 ml x 2) and was dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by alumina chromatography. Elution with chloroform/hexane (5/1) gave 12 mg (86%) of 18 as red solids. Mp = 245°C (decomp.). 1H NMR (500 MHz, CDCl3) δ = 3.44 (s, 6H), 4.95 (br, 1H), 6.83 (t, J = 7.7 Hz, 2H), 6.86 (d, J = 7.7 Hz, 4H), 7.17 (t, J = 7.7 Hz, 4H), 7.20 (s, 1H), 7.29 (t, J = 7.8 Hz, 2H), 7.51 (t, J = 7.8 Hz, 2H), 7.70 (d, J = 7.8 Hz, 2H), 8.18 (d, J = 7.8 Hz, 2H) ppm. 13C NMR (126 MHz, CDCl3) δ = 40.2, 115.6, 115.8, 119.8, 122.1, 123.9, 125.6, 127.8, 129.1, 145.0, 146.0, 148.1, 153.6 ppm. IR (KBr) νmax = 1495s, 1390s, 1108s, 750s cm–1. UV-vis (CH3CN) λmax = 226 (logε = 4.53), 235sh (4.50), 341 (4.59), 455 (4.03), 518sh (3.81) nm. MS (70 eV) m/z (%) = 479 (M++1, 37), 478 (100, M+), 372 (12), 371 (19), 357 (19), 356 (15). HRMS Calcd for C33H26N4 478.2158; found 478.2158.
3.9. 6,8-Dimethoxy-, 6,8-diethoxy-, and 6,8-di-n-butoxy- 1H-cyclohepta[2,1-b;3,4-b']diindoles (19–21)
- A mixture of 8 (27 mg, 0.080 mmol) and sodium methoxide (8.8 mg, 0.16 mmol) in 2 ml of methanol was heated at 130°C for 51 h in a sealed tube purged with argon. After being cooled to room temperature, the reaction mixture was poured into water (5 ml). Precipitates formed were filtered and washed with water (5 ml) and ethanol (3 ml). The solid obtained was purified by alumina column chromatography (eluted with 2/98 methanol/DCM), followed by silica gel column chromatography (eluted with 1/9 methanol/DCM), to give 13 mg (50%) of 19 as yellow powder. Mp = 280–281°C (decomp.). 1H NMR (500 MHz, DMSO-d6, 80°C) δ = 4.41 (s, 6H), 6.96 (s, 1H), 7.32 (t, J = 7.8 Hz, 2H), 7.49 (t, J = 7.8 Hz, 2H), 7.84 (d, J = 7.8 Hz, 2H), 8.50 (d, J = 7.8 Hz, 2H) ppm; 13C NMR (126 MHz, DMSO-d6, 80°C) δ = 56.2, 91.6, 113.8, 114.9, 120.6, 123.7, 125.3, 125.8, 142.6, 144.6, 164.3 ppm; IR (KBr) νmax = 1362s, 1298s cm–1; UV-vis (CH3CN) λmax = 223 (log ε = 4.48), 234sh (4.34), 272 (4.16), 307 (4.80), 321 (4.70), 333 (4.75), 373 (4.13), 429 (3.57) nm. MS (70 eV) m/z (%) = 328 (M+, 100), 281 (12),. 285 (14), 270 (10), 242 (15). HRMS Calcd for C21H16N2O2 328.1212; found 328.1207.Compounds 20–21 were synthesized similarly from 8 with sodium alkoxide in alcohol. 20: Orange prisms, mp = 277–278oC (decomp). 1H NMR (500 MHz, DMSO-d6, 90°C) δ = 1.70 (t, J = 6.9 Hz, 6H), 4.68 (q, J = 6.9 Hz, 4H), 6.96 (s, 1H), 7.33 (t, J = 7.6 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.85 (d, J = 7.6 Hz, 2H), 8.52 (d, J = 7.6 Hz, 2H) ppm [21]; 13C NMR (126 MHz, DMSO-d6, 90°C) δ = 14.2, 65.3, 93.3, 114.2, 114.9, 121.1, 123.9, 125.7, 125.9, 142.1, 144.2, 164.2 ppm; IR (KBr) νmax = 1583s, 1548s, 1413s, 1365vs, 1296s, 1243s, 1226s, 1209s, 1126s, 793s, 743s cm–1; UV-vis (CH3CN) λmax = 222 (logε = 4.34), 232sh (4.23), 272 (4.62), 307 (4.67), 320sh (4.56), 333 (4.61), 371 (4.00), 430 (3.41) nm; MS (70 eV) m/z (%) = 356 (M+, 100), 357 (26), 242 (15), 243 (11), 271 (21). HRMS Calcd for C23H20N2O2 356.1525; found. 356.1529.21: Orange microcrystals, mp = 245°C (decomp.). 1H NMR (500 MHz, DMSO-d6, 80°C) δ = 1.08 (t, J = 7.5 Hz, 6H), 1.69 (sextet, J = 7.8 Hz, 4H), 2.11 (quintet, J = 6.3 Hz, 4H), 4.65 (t, J = 6.3 Hz, 4H), 6.98 (s, 1H), 7.34 (t, J = 7.8 Hz, 2H), 7.50 (t, J = 7.8 Hz, 2H), 7.86 (d, J = 7.8 Hz, 2H), 8.53 (d, J = 7.8 Hz, 2H) ppm. 13C NMR (126 MHz, DMSO-d6, 120°C) δ = 13.0, 18.4, 30.3, 68.8, 92.8, 113.9, 114.9, 120.4, 123.4, 125.1, 125.8, 142.6, 144.5, 163.7 ppm. IR (KBr) νmax = 1365s, 1302s, 746s cm–1. UV-vis (DMSO) λmax = 311 (logε = 4.72), 337 (4.67), 373 (4.10), 438 (3.55) nm. MS (70 eV) m/z (%) = 412 (100, M+), 300 (26), 271 (16), 243 (11). HRMS Calcd for C27H28N2O2 412.2151; found 412.2156.
3.10. 6-Chloror-8-ethoxy-1H-cyclohepta[2,1-b;3,4-b'] -diindole (22)
- A mixture of 8 (34 mg, 0.10 mmol) and sodium ethoxide (6.8 mg, 0.10 mmol) in 1 ml of ethanol was heated at 130°C for 39 h in a sealed tube purged by argon. After being cooled to room temperature, the reaction mixture poured into water (5 ml) and the precipitate formed was filtered off. The solid obtained was purified by alumina column chromatography. Elution with methanol/dichloromethane (10/90) gave 24 mg (69%) of 22 as orange solids. Mp = 292– 294°C. 1H NMR (500 MHz, DMSO-d6, 100°C) δ = 1.68 (t, J = 6.9 Hz, 3H), 4.64 (q, J = 6.9 Hz, 2H), 7.31 (s, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.43 (t, J = 7.8 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 8.56 (d, J = 7.8 Hz, 1H), 8.82 (d, J = 7.8 Hz, 1H) ppm [21]; 13C NMR (126 MHz, DMSO-d6, 100°C) δ = 14.4, 65.9, 107.8, 113.7, 115.0, 118.0, 121.6, 121.9, 122.4, 124.3, 124.6, 124.9, 126.9, 127.4, 127.7, 128.6, 140.7, 141.1, 142.1, 145.7, 161.4 ppm; IR (KBr) νmax = 1574s, 1557s, 1531s, 1411s, 1395vs, 1372vs, 1270vs, 1262vs, 1234s, 1213s, 1196vs, 1114s, 863s, 806s, 754vs, 743vs cm–1; UV-vis (CH3CN) λmax = 221 (logε = 4.41), 232sh (4.26), 263sh (3.94), 316 (4.65), 335 (4.64), 354sh (4.23), 372sh (3.91), 389 (3.75), 413 (3.51), 470 (3.66) nm; MS (70 eV) m/z (%) = 348 (M+, 35), 346 (M+, 100), 318 (28), 291 (10), 289 (28), 283 (11), 255 (21), 254 (11), 253 (15). HRMS Calcd for C21H1535ClN2O; 346.0873; found 346.0868.
3.11. 6-Ethoxy-8-piperidino-1H-cyclohepta[2,1-b;3,4-b'] -diindole (23)
- A mixture of 22 (24 mg, 0.069 mmol), piperidine (7 μl, 6 mg, 0.07 mmol) and triethylamine (10 μl, 7 mg, 0.07 mmol) in 1.5 ml of dry DMF was heated at 110°C for 1 h under argon atmosphere. After being cooled to room temperature, the reaction mixture was poured into water (5 ml) and precipitates formed were filtered off. The solid obtained was purified by alumina column chromatography. Elution with methanol/dichloromethane (3/97) gave 25 mg (93%) of 23 as yellow solids. Mp = 260–263°C. 1H NMR (500 MHz, CDCl3, 50°C) δ = 1.74 (t, J = 6.9 Hz, 3H), 1.86–1.99 (m, 4H), 3.04 (br, 2H), 3.67 (br, 2H), 4.45 (q, J = 6.9 Hz, 2H), 6.95 (s, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 7.7 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H), 8.54 (d, J = 7.7 Hz, 1H), 8.61 (d, J = 7.7 Hz, 1H) ppm [21]; 1H NMR (500 MHz, DMSO-d6, 100°C) δ = 1.68 (t, J = 6.8 Hz, 3H), 1.73 (br, 2H), 1.89 (br, 4H), 3.42 (br, 4H), 4.63 (q, J = 6.8 Hz, 2H), 7.05 (s, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.7 Hz, 1H), 7.46 (t, J = 7.7 Hz, 2H), 7.82 (d, J = 7.7 Hz, 2H), 8.49 (d, J = 7.7 Hz, 1H), 8.52 (d, J = 7.7 Hz, 1H) ppm [21]; 13C NMR (126 MHz, CDCl3, 50°C) δ = 14.9, 24.4, 26.1, 54.4, 65.0, 100.2, 115.3, 115.4, 116.5, 118.4, 120.8, 121.3, 123.1, 124.4, 125.9, 126.1, 126.5, 126.6, 143.9, 144.1, 144.2, 145.3, 160.3, 163.7 ppm; IR (KBr) νmax = 1559s, 1540s, 1410s, 1361s, 1226s, 1203s, 749s, cm–1; UV-vis (CH3CN) λmax = 217 (logε = 4.80), 234sh (4.66), 319sh (4.72), 335 (4.89), 392 (4.16), 418 (4.11), 462sh (3.49) nm; MS (70 eV) m/z (%) = 395 (M+, 100), 366 (15), 284 (12), 283 (11), 255 (17). HRMS Calcd for C26H25N3O 395.1998; found 395.1991.
3.12. 7-Piperidino-1H-cyclohepta[2,1-b;3,4-b']diindole (24, Nu = piperidino)
- A mixture of 5 (50 mg, 0.17 mmol) and piperidine (1.0 ml) was heated at 130°C for 62 h in a sealed tube purged by nitrogen. After being cooled to room temperature, the reaction mixture was poured into a saturated sodium bicarbonate solution (20 ml) and was extracted with DCM (30 x 3 ml). The combined organic layer was washed with brine (25 ml) and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (eluted with ethanol/chloroform, 7/93), followed by alumina column chromatography (eluted with ethyl acetate/chloroform, 50/50) to give 9 mg (16%) of 24 as dark purple solids. Mp > 200°C (decomp.). 1H NMR (500 MHz, CDCl3) δ = 1.73 (quin, J = 5.3 Hz, 2H), 1.95 (quin, J = 5.3 Hz, 4H), 3.41 (t, J = 5.3 Hz, 4H), 7.39 (t, J = 7.7 Hz, 2H), 7.51 (t, J = 7.7 Hz, 2H), 7.56 (d, J = 7.7 Hz, 2H), 8.31 (d, J = 7.7 Hz, 2H), 8.64 (s, 2H) ppm [21]; 13C NMR (126 MHz, CDCl3) δ = 24.1, 26.7, 54.5, 115.6, 120.0, 121.0, 124.7, 127.2, 128.4, 130.2, 142.4, 146.6, 148.0 ppm; IR (KBr) νmax =1496s, 1410s, 1366s, 1196s, 1110s, 744s cm–1; UV-vis (CH3CN) λmax = 225 (logε = 4.30), 234sh (4.28), 278 (4.17), 318 (4.57), 331 (4.52), 347 (4.15), 362sh (4.05), 381sh (4.02), 535 (3.61) nm; MS (70 eV) m/z (rel int.) = 351 (M+, 100), 352 (27), 266 (12), 267 (10), 268 (22), 295 (10), 349 (12), 350 (19). HRMS Calcd for C24H21N3, 351.1735; found 351.1738.
3.13. X-ray Crystallographic Analysis of 14
- Diffraction measurements were conducted using a Rigaku R-AXIS RAPID diffractometer at –100°C. Crystal data for 14 are as follows; monoclinic, space group; C2/c (# 15), a; 25.1397(5) Å, b; 10.4773(2) Å, c; 21.4999(4) Å, β; 123.6023(7)°, V; 4716.7(2) Å3, Z; 8, R; 0.0454, wR2; 0.1054, R1; 0.0418 (I>2.00σ(I)), and S; 1.081. Tables of fractional atomic coordinates, thermal parameters, bond lengths, and angles have been deposited at the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom (CCDC 1427913) [Direct line: +44 1223 762910, Fax: +44 (0) 1233 336033, e-mail: deposit@ccdc.cam.ac.uk].
4. Conclusions
- We have demonstrated that the halogenated compounds 5–8 could be synthesized from 2,2’-biindole (9) and studied on their nucleophilic substitution reactions. While 8 undergoes the substitution reaction with amine and alcohol to afford the various 6,8-substituted products, compounds 5–7 showed rather reluctance to undergo substitution reactions. Again, it should be addressed that many kinds of the 6,8-disubstituted derivatives can be synthesized from 8 by the method developed in this study. The difference in the reactivity between 5–7 and 8 was rationalized by the relative stability of the expected anionic intermediates based on the results of DFT calculations. Further derivatization at the halogenated positions of 5–8 by metal-catalyzed couplings with boronic acids, organozincates and stannanes is now in progress.
ACKNOWLEDGEMENTS
- Financial support from the Faculty of Science in Shinshu University (for M.O.) is greatly acknowledged.
Supplementary Material
- Output files for computations reported in this work are available upon request from the corresponding author.