-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2016; 6(2): 54-80
doi:10.5923/j.ajoc.20160602.02

Design, Synthesis, and Biological Evaluation of Novel 5-Substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles as Potent Antioxidants
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAmgad M. Rabie , Atif S. Tantawy , Sahar M. I. Badr
Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
Correspondence to: Amgad M. Rabie , Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A novel series of 5-(5-substituted-1,3,4-oxadiazol-2-yl)benzene-1,2,3-triols (3n-z) was designed, synthesized, and evaluated for its potential antioxidant activities. Structural modifications at position 5 of the 1,3,4-oxadiazole scaffold (linked to a fixed antioxidant 3,4,5-trihydroxyphenyl moiety at position 2 of the ring) was expected to give new 1,3,4-oxadiazole derivatives with a wide spectrum of biological antioxidant activities. Undoubted elucidation and full confirmation of the chemical structures of all the newly synthesized compounds were accomplished using the spectroscopical and elemental analyses. The pharmacological screening for evaluation of the antioxidant activity of these new thirteen target 5-substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles (3n-z) was done by using two of the most common in vitro antioxidant assays. The results of both assays showed that compounds 3w,s,u (the fumaric, malonic, and citric acids-derived 1,3,4-oxadiazoles, respectively) surprisingly exhibited very high and significant antioxidant activities, and they could be very promising lead and parent compounds for the design and synthesis of new antioxidant agents by further in vivo biological evaluations, structural modifications, and computational studies.
Keywords: 1,3,4-Oxadiazoles, Phenolic hydroxyl groups, Microwave-assisted synthesis, Reactive oxy(nitro)gen species, Antioxidant activities
Cite this paper: Amgad M. Rabie , Atif S. Tantawy , Sahar M. I. Badr , Design, Synthesis, and Biological Evaluation of Novel 5-Substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles as Potent Antioxidants, American Journal of Organic Chemistry, Vol. 6 No. 2, 2016, pp. 54-80. doi: 10.5923/j.ajoc.20160602.02.
Article Outline
1. Introduction
- Antioxidants are chemical compounds that may protect cells from the damage caused by unstable molecules known as free radicals; these antioxidant substances include those of a nonenzymatic as well as an enzymatic nature [1-4]. The oxidative stress and damage, caused by the attack of excess free radicals and other ROS (reactive oxygen species)/RNS (reactive nitrogen species) (i.e., other nonradicals), is implicated in the pathogenesis and development (and also indicative) of various diseases in human, i.e., either as a primary cause or as a consequence of disease progression, specially, the chronic diseases and degenerative disorders [2,5-7], such as neurodegenerative diseases [2,3,5-14], cardiovascular diseases [2,3,5,6,8,10,14-16], hepatic and pancreatic diseases [2,3,5-8,10,14,16-18], renal and urological diseases [2,5,6,8,10,14], respiratory diseases [2,3,6,8,10], ocular (ophthalmic) diseases [2,3,5,6,8,10,14], dermal/hair/nails diseases [2,3,6,8,19], orthopaedic diseases [2,3,5,6,10], hematologic diseases [2,3,6,8,14], gastrointestinal (gastroenterological) or digestive diseases [2,3,6,8,10], immunological and infectious diseases [2,3,6], otorhinolaryngological (ear, nose, and throat) and dental diseases [2,3,5,8,10], andrological/gynecological/obstetrical (reproductive system) diseases [2,3,6,8], and other (e.g., multiorgan or multisystem) diseases [2,5-10,14,16-18]. Antioxidants can be classified according to many items [3,7,8,15], but, generally, they can be classified into natural antioxidants (including many enzymes such as superoxide dismutase and glutathione reductase; some vitamins such as vitamins E and C; carotenoids such as carotenes and lycopene; some polyphenols such as resveratrol and silymarin; some hormones such as melatonin; some coenzymes such as ubiquinol which is the reduced form of coenzyme Q10; some inorganic nutrients/chemical elements such as selenium and copper; and various natural antioxidant compounds such as glutathione, bilirubin, and uric acid) and synthetic antioxidants (including many dietary or nutritional antioxidant supplements such as ebselen, trolox, disufenton sodium, and raxofelast; many food additives and preservatives such as propyl gallate and butylated hydroxytoluene; and other synthetic antioxidant medicines) [2-4,6-8,14]. Figure 1 (below) shows the chemical structures of two of the most potent antioxidants (vitamin C and trolox).
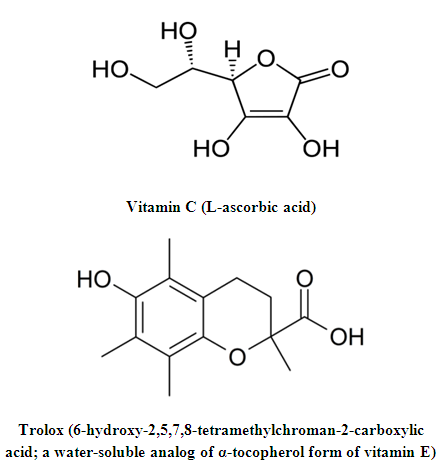 | Figure 1. The chemical structures of vitamin C (a very strong natural antioxidant) and trolox (a strong synthetic antioxidant) |
 | Figure 2. The chemical structure of the new antioxidant 5-(fur-2-yl)-2- (3,4,5-trihydroxyphenyl)-1,3,4-oxadiazole (3Oxa) synthesized by D. K. Mehta and R. Das |
 | Figure 3. The Proposed general antioxidant 2,5-disubstituted-1,3,4-oxadiazole model |
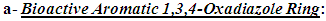 Many of the compounds containing the bioactive 1,3,4-oxadiazole ring (the scaffold and the main part in this model) efficiently exhibit electron donor-acceptor properties, specially when an EDG (electron-donating group) is attached to this ring, as the introduction of EDGs into the electron-withdrawing heterocyclic 1,3,4-oxadiazole ring affords excellent electron donor-acceptor compounds that are easily both oxidized and reduced (i.e., having excellent antioxidant activities by being easily oxidized by oxidants which include ROS/RNS and all other free radicals) [21]. 1,3,4-oxadiazole moiety traps free radicals and ROS/RNS by potential conjugation of the aromatic structure, in addition, it is characterized by many unique properties that are not collectively present in most other ring systems, such as acting as a hydrogen-binding domain (this greatly increases the antioxidant properties of the compounds containing it) [20-22]. All the useful properties of 1,3,4-oxadiazole ring collectively have dramatic aiding effects on the net antioxidant biological activities of the ring derivatives and this helps to reach the optimal levels.
Many of the compounds containing the bioactive 1,3,4-oxadiazole ring (the scaffold and the main part in this model) efficiently exhibit electron donor-acceptor properties, specially when an EDG (electron-donating group) is attached to this ring, as the introduction of EDGs into the electron-withdrawing heterocyclic 1,3,4-oxadiazole ring affords excellent electron donor-acceptor compounds that are easily both oxidized and reduced (i.e., having excellent antioxidant activities by being easily oxidized by oxidants which include ROS/RNS and all other free radicals) [21]. 1,3,4-oxadiazole moiety traps free radicals and ROS/RNS by potential conjugation of the aromatic structure, in addition, it is characterized by many unique properties that are not collectively present in most other ring systems, such as acting as a hydrogen-binding domain (this greatly increases the antioxidant properties of the compounds containing it) [20-22]. All the useful properties of 1,3,4-oxadiazole ring collectively have dramatic aiding effects on the net antioxidant biological activities of the ring derivatives and this helps to reach the optimal levels.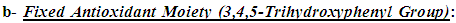 It was very important in the design of this antioxidant model to have a fixed antioxidant moiety at any carbon atom of the two carbons (i.e., at position 2) of the 1,3,4-oxadiazole ring that is not changed through all the compounds of this series (1,3,4-oxadiazoles series) to establish the main moiety responsible for the occurrence of the principal in vivo redox cycle in which the oxidized form of each 1,3,4-oxadiazole derivative is much more stable (i.e., favorable and predominant) than ROS/RNS and other free radicals (i.e., than most active in vivo oxidants). This group moiety was chosen to have electronegative heteroatoms that are similar to the centered electronegative heteroatom of the heterocyclic 1,3,4-oxadiazole ring (i.e., oxygen (O) atoms), and as a result, 3,4,5-trihydroxyphenyl group was chosen for this 1,3,4-oxadiazoles series. The polyhydroxyphenolic moiety (i.e., the phenolic hydroxyl (OH) groups) has very strong antioxidant properties as it characterizes by donating its hydrogens (or, first, giving an electron, then, the proton) to any oxidant or radical, to catch it, very easily (i.e., it is a very good hydrogen donor) [22-25].
It was very important in the design of this antioxidant model to have a fixed antioxidant moiety at any carbon atom of the two carbons (i.e., at position 2) of the 1,3,4-oxadiazole ring that is not changed through all the compounds of this series (1,3,4-oxadiazoles series) to establish the main moiety responsible for the occurrence of the principal in vivo redox cycle in which the oxidized form of each 1,3,4-oxadiazole derivative is much more stable (i.e., favorable and predominant) than ROS/RNS and other free radicals (i.e., than most active in vivo oxidants). This group moiety was chosen to have electronegative heteroatoms that are similar to the centered electronegative heteroatom of the heterocyclic 1,3,4-oxadiazole ring (i.e., oxygen (O) atoms), and as a result, 3,4,5-trihydroxyphenyl group was chosen for this 1,3,4-oxadiazoles series. The polyhydroxyphenolic moiety (i.e., the phenolic hydroxyl (OH) groups) has very strong antioxidant properties as it characterizes by donating its hydrogens (or, first, giving an electron, then, the proton) to any oxidant or radical, to catch it, very easily (i.e., it is a very good hydrogen donor) [22-25].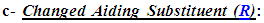 An important part, which has a complementary role in increasing the antioxidant activities of this model, is the changeable aiding moiety or substituent
An important part, which has a complementary role in increasing the antioxidant activities of this model, is the changeable aiding moiety or substituent 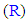 at position 5 (i.e., at the other carbon) of the 1,3,4-oxadiazole scaffold. The main function of this aiding substituent
at position 5 (i.e., at the other carbon) of the 1,3,4-oxadiazole scaffold. The main function of this aiding substituent 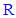 is to increase the net total antioxidant activities of the target compounds, directly, through helping their pharmacodynamic properties (i.e., through giving an additive antioxidant effect to the activity of the original parent 2-(3,4,5-trihydroxyphenyl)-1,3, 4-oxadiazole compound and/or aiding the mechanism of the antioxidant action of the original parent 2-(3,4, 5-trihydroxyphenyl)-1,3,4-oxadiazole compound) and/or, indirectly, through helping their pharmacokinetic parameters to reach the required optimal values according to the need, target site(s) of administration, and target human organs (e.g., by increasing their lipophilicity/ hydrophilicity, rate of absorption, and bioavailability). The chemical structures of all the
is to increase the net total antioxidant activities of the target compounds, directly, through helping their pharmacodynamic properties (i.e., through giving an additive antioxidant effect to the activity of the original parent 2-(3,4,5-trihydroxyphenyl)-1,3, 4-oxadiazole compound and/or aiding the mechanism of the antioxidant action of the original parent 2-(3,4, 5-trihydroxyphenyl)-1,3,4-oxadiazole compound) and/or, indirectly, through helping their pharmacokinetic parameters to reach the required optimal values according to the need, target site(s) of administration, and target human organs (e.g., by increasing their lipophilicity/ hydrophilicity, rate of absorption, and bioavailability). The chemical structures of all the  groups of the thirteen target compounds (3n-z) are demonstrated in Table 1.In view of the above-mentioned introductory facts, it is concluded that 1,3,4-oxadiazole scaffold and 1,2,3-trihydroxyphenyl (3,4,5-trihydroxyphenyl) moiety have been known to have antioxidant properties and, therefore, according to ‘‘the combination principles’’, if an aromatic 1,3,4-oxadiazole ring is directly linked with a 3,4,5-trihydroxyphenyl group at position 2 of the ring and with an aiding group at position 5 of the ring, the produced 2,5-disubstituted-1,3,4-oxadiazoles (1,3,4-oxadiazole scaffold derivatives) should be or are expected to be capable of scavenging free radicals, ROS, RNS, and all other types of oxidants in a potent ideal manner (i.e., potent antioxidants).
groups of the thirteen target compounds (3n-z) are demonstrated in Table 1.In view of the above-mentioned introductory facts, it is concluded that 1,3,4-oxadiazole scaffold and 1,2,3-trihydroxyphenyl (3,4,5-trihydroxyphenyl) moiety have been known to have antioxidant properties and, therefore, according to ‘‘the combination principles’’, if an aromatic 1,3,4-oxadiazole ring is directly linked with a 3,4,5-trihydroxyphenyl group at position 2 of the ring and with an aiding group at position 5 of the ring, the produced 2,5-disubstituted-1,3,4-oxadiazoles (1,3,4-oxadiazole scaffold derivatives) should be or are expected to be capable of scavenging free radicals, ROS, RNS, and all other types of oxidants in a potent ideal manner (i.e., potent antioxidants).2. Results and Discussion
2.1. Chemical Synthesis
- The synthetic scheme of this research (Scheme 1, illustrated below) was adopted for the synthesis of the target compounds of the 1,3,4-oxadiazole series (compounds 3n-z). Ethyl 3,4,5-trihydroxybenzoate (ethyl gallate, 1nz) is the first intermediate compound synthesized in order to reach the final target compounds 3n-z (it is an old ester which is conventionally synthesized from 3,4, 5-trihydroxybenzoic or gallic acid by direct esterification), while the second intermediate compound is 3,4, 5-trihydroxybenzohydrazide (galloyl hydrazide, 2nz) which is either conventionally synthesized from the corresponding ester 1nz, or directly synthesized from the corresponding carboxylic acid (i.e., gallic acid) by MW-assisted (microwave-assisted) method without passing through the esterification step. The final step in this scheme is the oxidative cyclodehydration reaction which is used for the synthesis of all the different new thirteen target 5-(5-substituted-1,3,4-oxadiazol-2-yl)benzene-1,2,3-triols (5-substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles, final products 3n-z). All these synthetic steps are discussed below in details.
|
 of all the diverse
of all the diverse  substituents present in the target 1,3,4-oxadiazoles (3n-z)
substituents present in the target 1,3,4-oxadiazoles (3n-z) 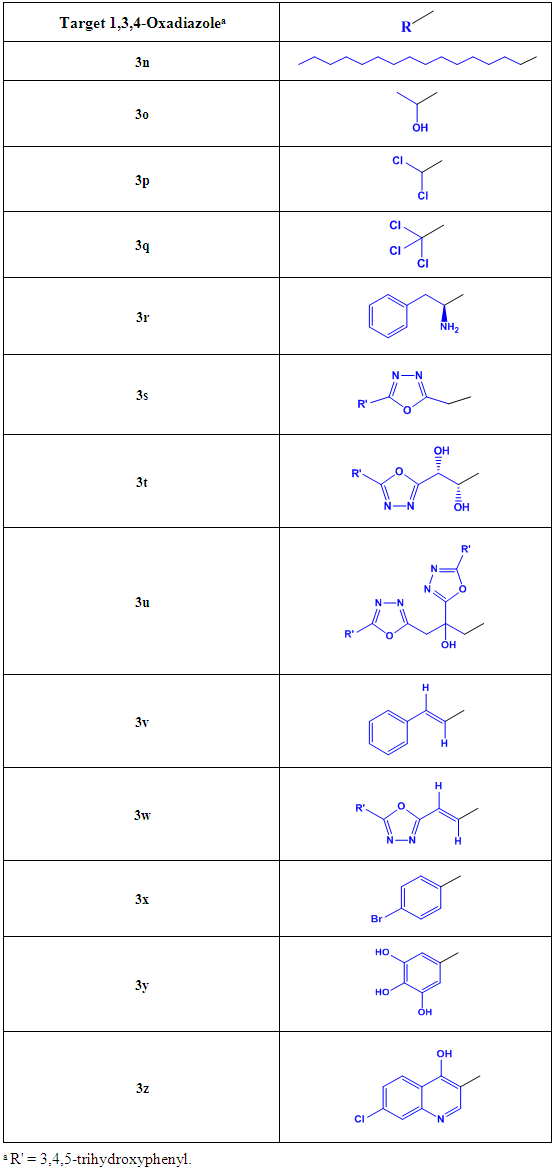
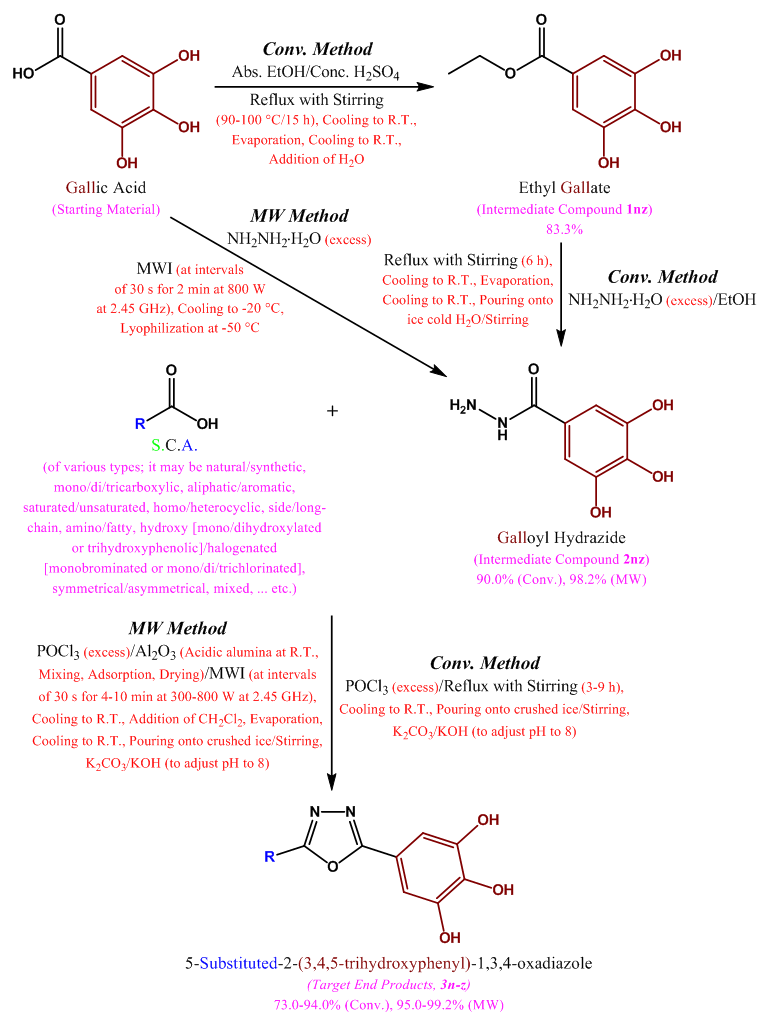 | Scheme 1. Synthesis of the target 5-(5-substituted-1,3,4-oxadiazol-2-yl)benzene-1,2,3-triols (3n-z) |
2.1.1. Conventional Synthesis of Ethyl Gallate (1nz)
- 1nz was synthesized by using the normal conventional esterification reaction of anhydrous gallic acid with absolute EtOH (ethanol) as the alcohol and Conc. H2SO4 (concentrated sulfuric acid) as the catalyst (strong dehydrating agent). EtOH acts, also, as the refluxing solvent for this reaction at about 90-100 °C for 15 h (hours). This reaction was reported by many scientists and researchers like Shigeki Takaoka et al. [26] and Krishna C. Majumdar et al. [27] (all in 2009). There are some other methods for the synthesis of 1nz from gallic acid, e.g., the direct method of Ambika et al. [28] and the indirect method of Kosuke Dodo et al. [29] (both methods were reported in 2008), but they are expensive, complicated, and less common.
2.1.2. Synthesis of Galloyl Hydrazide (2nz)
- 2nz was synthesized by using the hydrazinolysis reaction which is suggested to proceed via the normal nucleophilic substitution mechanism [30] in which NH2NH2 (hydrazine) molecule, being a good nucleophile, attacks the electrophilic carbon of the carbonyl group of either the corresponding ester (1nz) or the corresponding carboxylic acid (gallic acid). This synthetic step was achieved in this current research through either one of the following two methods:
 It is the known traditional method for the synthesis of 2nz in which the corresponding ester (mainly, the ethyl ester or 1nz) is hydrazinolyzed using NH2NH2.H2O (hydrazine hydrate) in the presence of EtOH as the refluxing solvent for the reactants (for 6 h in this current research). Generally, many of the references that reported this method in the literature mentioned the use of the ethyl ester for this reaction [22,31-38], some others mentioned the use of the propyl ester [39-46], while few others mentioned the use of the methyl ester [33,47-50].The ethyl ester 1nz was preferred, in this research, over the famous propyl gallate ester (and all other gallate esters in general), i.e., it is the ester of choice for this hydrazinolysis reaction, due to many reasons, such as providing the reaction with an internal autosolvent through giving EtOH as a byproduct of the reaction which could be condensed, recovered, and reused as a solvent for the reaction mixture, so less volume of the primary original EtOH would be used leading to reduced reaction costs, unlike the propyl ester which would give propanol instead; also ethyl group is a much better leaving group than the longer propyl group in this reaction, because it is easier to leave than the propyl one which is heavier than it, so a faster reaction with better yield would be obtained; and, in addition to these advantages of using it over the propyl gallate, the ethyl ester is mostly the first common choice, for this type of reactions (i.e., for condensation reactions).
It is the known traditional method for the synthesis of 2nz in which the corresponding ester (mainly, the ethyl ester or 1nz) is hydrazinolyzed using NH2NH2.H2O (hydrazine hydrate) in the presence of EtOH as the refluxing solvent for the reactants (for 6 h in this current research). Generally, many of the references that reported this method in the literature mentioned the use of the ethyl ester for this reaction [22,31-38], some others mentioned the use of the propyl ester [39-46], while few others mentioned the use of the methyl ester [33,47-50].The ethyl ester 1nz was preferred, in this research, over the famous propyl gallate ester (and all other gallate esters in general), i.e., it is the ester of choice for this hydrazinolysis reaction, due to many reasons, such as providing the reaction with an internal autosolvent through giving EtOH as a byproduct of the reaction which could be condensed, recovered, and reused as a solvent for the reaction mixture, so less volume of the primary original EtOH would be used leading to reduced reaction costs, unlike the propyl ester which would give propanol instead; also ethyl group is a much better leaving group than the longer propyl group in this reaction, because it is easier to leave than the propyl one which is heavier than it, so a faster reaction with better yield would be obtained; and, in addition to these advantages of using it over the propyl gallate, the ethyl ester is mostly the first common choice, for this type of reactions (i.e., for condensation reactions). It is a newly discovered and designed solventless method as it needs only the two reactants, gallic acid and NH2NH2.H2O, with no need for any solvents, catalysts, inert supports, or other reagents [35]. Here, the reaction is directly made on the corresponding original carboxylic acid (gallic acid) without passing through the first step of ester synthesis, so it is just one fast greener step (unlike the previous conventional method which includes two slow hazardous separate steps, firstly, to form the ester 1nz from gallic acid, then to form the hydrazide 2nz from the ester 1nz) with very high excellent rates of saving time, energy, and money (in addition, it is environmentally safe) relative to the traditional method of conventional heating (Table 2 below shows a detailed comparison between both the conventional and MW methods for 2nz synthesis beginning from gallic acid).
It is a newly discovered and designed solventless method as it needs only the two reactants, gallic acid and NH2NH2.H2O, with no need for any solvents, catalysts, inert supports, or other reagents [35]. Here, the reaction is directly made on the corresponding original carboxylic acid (gallic acid) without passing through the first step of ester synthesis, so it is just one fast greener step (unlike the previous conventional method which includes two slow hazardous separate steps, firstly, to form the ester 1nz from gallic acid, then to form the hydrazide 2nz from the ester 1nz) with very high excellent rates of saving time, energy, and money (in addition, it is environmentally safe) relative to the traditional method of conventional heating (Table 2 below shows a detailed comparison between both the conventional and MW methods for 2nz synthesis beginning from gallic acid).
2.1.3. Synthesis of 5-Substituted-2-(3,4,5-trihydroxyphe- nyl)-1,3,4-oxadiazoles (3n-z)
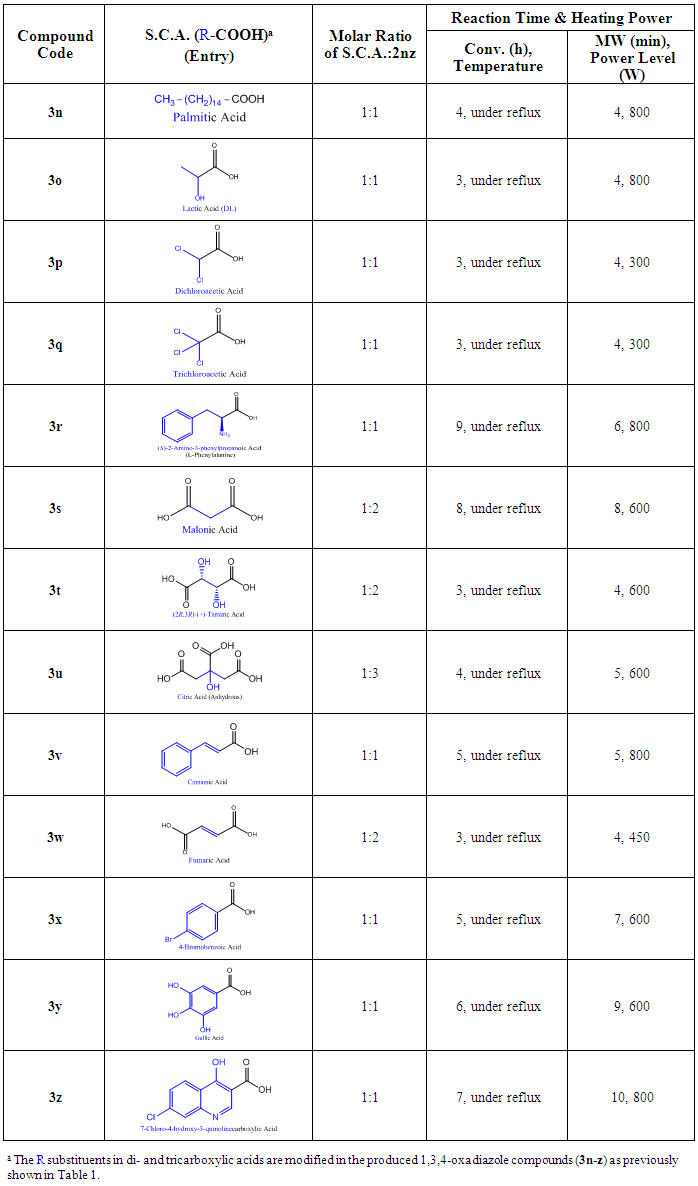
- Compounds 3n-z were synthesized by using the oxidative cyclodehydration reaction of the hydrazide 2nz with various starting carboxylic acids (S.C.A.) in POCl3 (phosphorus oxychloride) as the strong dehydrating agent, which is either refluxing POCl3 (conventional method) or adsorbed POCl3 (MW-assisted method) [22,31,32,34,39,47,50,53,54]. Table 3 shows the heating time (in h and min “minutes”, respectively) and power (in W “watts”) needed by the conventional method (method A) and MW method (method B). All compounds were synthesized in good to excellent yields ranging from 73.0 to 94.0% (conventionally) and from 95.0 to 99.2% (MW method) (see the Experimental Work for details). The designed MW method, used here, gave much better results than the conventional one (see the detailed table, Table 4, for the comparison of MW method versus conventional one for the synthesis of 2,5-disubstituted-1,3,4-oxadiazoles from their precursors, 2nz and S.C.A.).
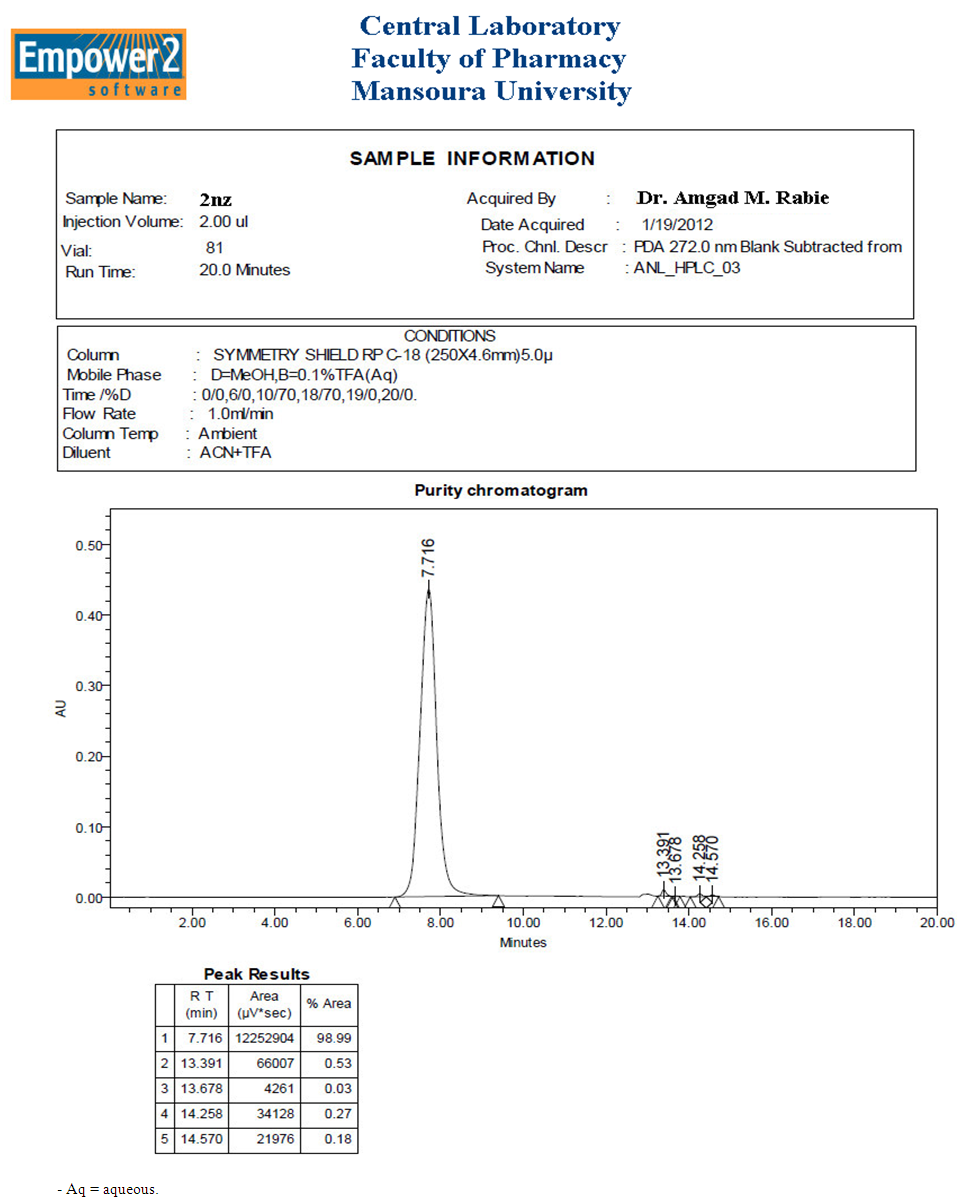 | Figure 4. A scanned copy of the HPLC chart of analysis of compound 2nz obtained in this work (including sample information, analytical conditions, purity chromatogram, and peak results) |
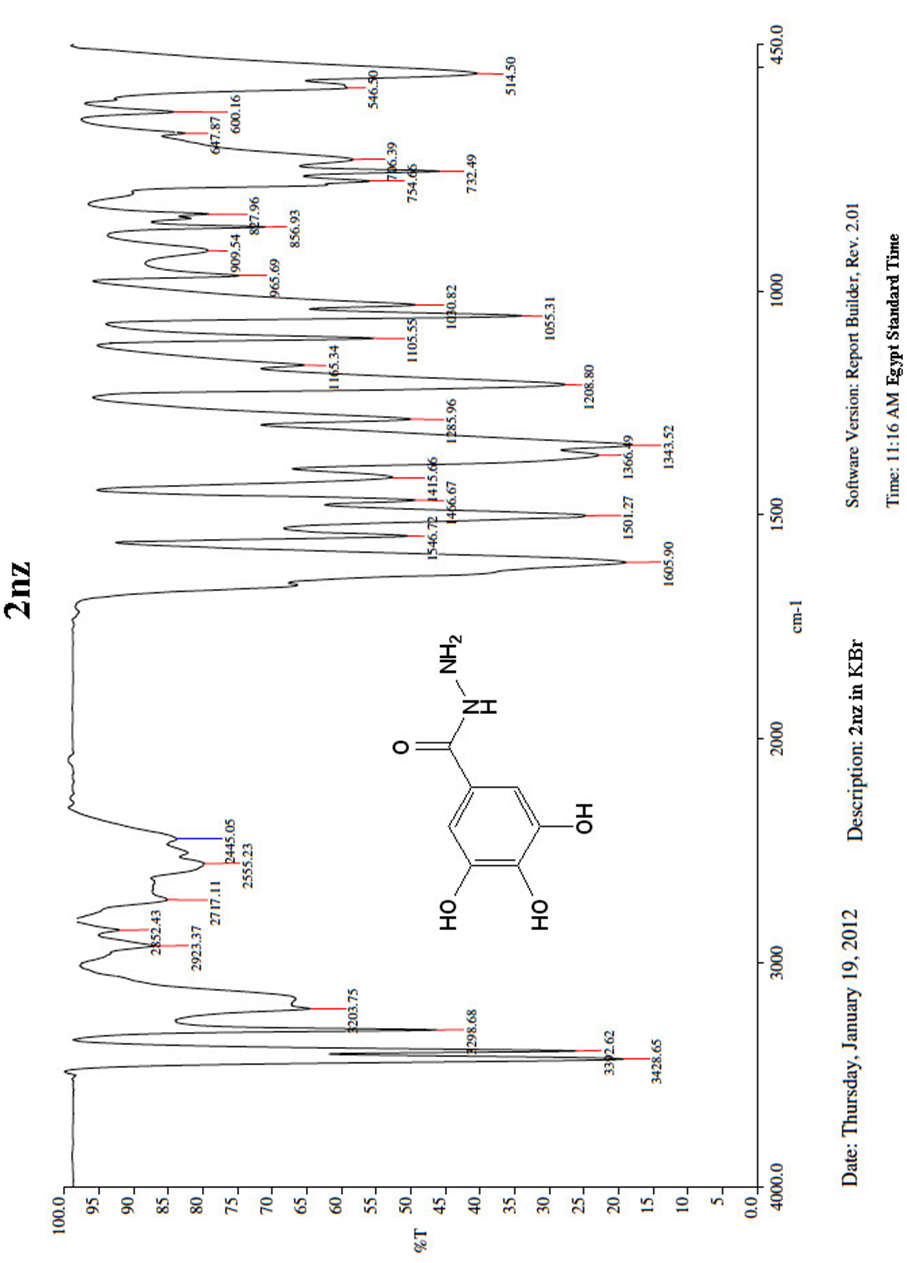 | Figure 5. A scanned copy of the spectral chart obtained upon IR spectroscopical analysis of the sample of the compound 2nz |
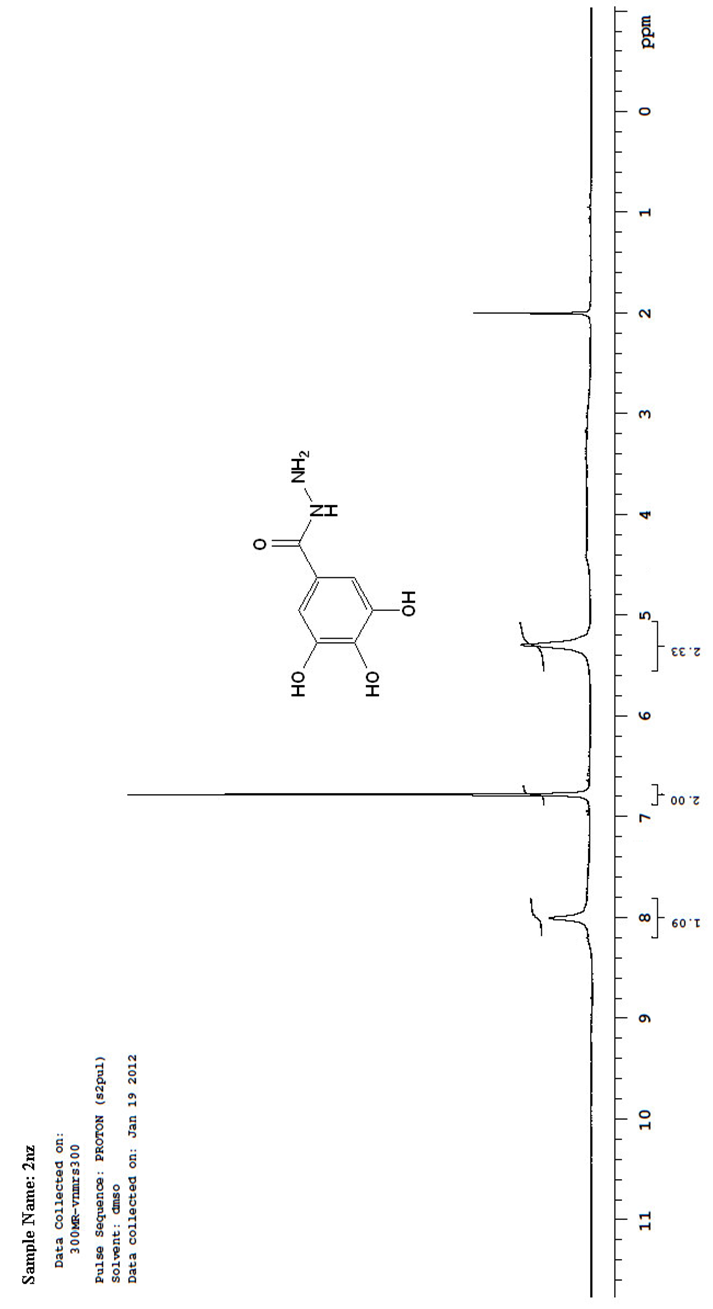 | Figure 6. A scanned copy of the spectral chart obtained upon 1H-NMR spectroscopical analysis of the sample of the compound 2nz |
 | Figure 7. A scanned copy of the spectral chart (explained) obtained upon 13C-NMR spectroscopical analysis of the sample of the compound 2nz |
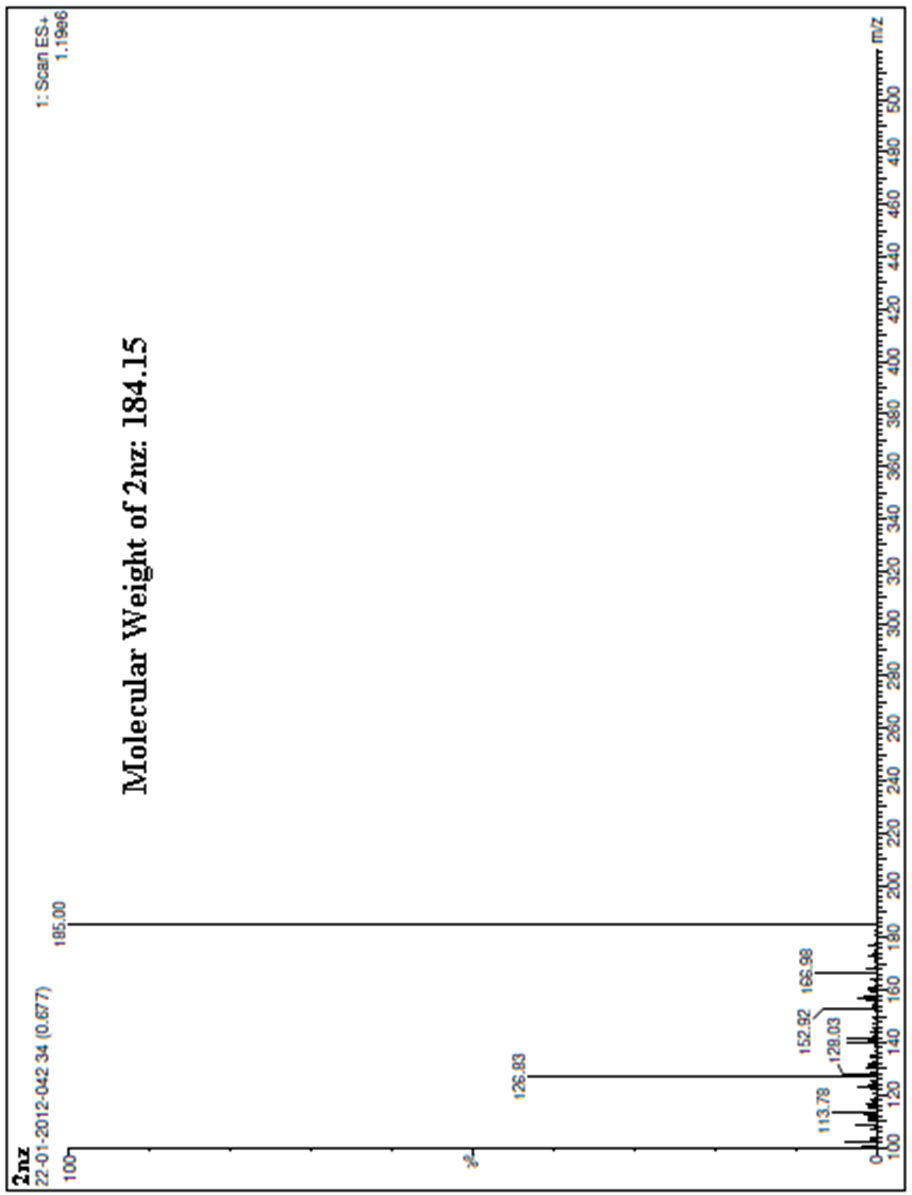 | Figure 8. A scanned copy of the spectral chart obtained upon MS analysis of the sample of the compound 2nz |
|
|
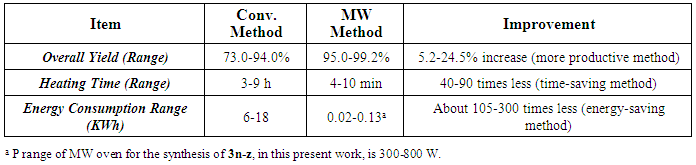
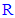 groups that are attached to position 5 of the 1,3,4-oxadiazole ring in each compound in this series (3n-z) were specific confirmations for the chemical structure of each compound [52]. In 1H-NMR spectra, mainly, the clear absence of any characteristic signal for the proton of the OH group of any carboxyl moiety in the range of 10.5-15.0 ppm (as all the 1H-NMR spectral charts resulted from the analysis of compounds 3n-z contain no signals in this large region of the spectrum at all) was an excellent confirmation of conversion of all the carboxylic acids (through reacting with 2nz) to the substituted 1,3,4-oxadiazole heteroring, furthermore, the general presence of the characteristic singlet signals at 5.333-5.548 ppm indicated the existence of the three protons of the three adjacent phenolic hydroxyl groups attached to a benzene ring (its existence and attachment to position 2 of the formed 1,3,4-oxadiazole ring was confirmed by the presence of the common characteristic singlet signals of the only two protons of this phenyl ring at 6.716-7.313 ppm in all the produced compounds of this 1,3,4-oxadiazole series), and, finally, the other varied signals representing the protons (if present) of specific different
groups that are attached to position 5 of the 1,3,4-oxadiazole ring in each compound in this series (3n-z) were specific confirmations for the chemical structure of each compound [52]. In 1H-NMR spectra, mainly, the clear absence of any characteristic signal for the proton of the OH group of any carboxyl moiety in the range of 10.5-15.0 ppm (as all the 1H-NMR spectral charts resulted from the analysis of compounds 3n-z contain no signals in this large region of the spectrum at all) was an excellent confirmation of conversion of all the carboxylic acids (through reacting with 2nz) to the substituted 1,3,4-oxadiazole heteroring, furthermore, the general presence of the characteristic singlet signals at 5.333-5.548 ppm indicated the existence of the three protons of the three adjacent phenolic hydroxyl groups attached to a benzene ring (its existence and attachment to position 2 of the formed 1,3,4-oxadiazole ring was confirmed by the presence of the common characteristic singlet signals of the only two protons of this phenyl ring at 6.716-7.313 ppm in all the produced compounds of this 1,3,4-oxadiazole series), and, finally, the other varied signals representing the protons (if present) of specific different 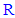 groups that are attached to position 5 of the 1,3,4-oxadiazole ring in each compound in this series (3n-z) were specific confirmations for the chemical structure of most compounds among compounds 3n-z (only those whose
groups that are attached to position 5 of the 1,3,4-oxadiazole ring in each compound in this series (3n-z) were specific confirmations for the chemical structure of most compounds among compounds 3n-z (only those whose 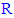 group has protons in its structure) [52]. The specific values of MS and elemental analyses (see the Experimental Work for details) gave confirmatory assignments and further final evidences for the characterization of the structures of all these newly synthesized compounds (3n-z) [52].
group has protons in its structure) [52]. The specific values of MS and elemental analyses (see the Experimental Work for details) gave confirmatory assignments and further final evidences for the characterization of the structures of all these newly synthesized compounds (3n-z) [52]. 2.2. Biological Evaluation
- The in vitro antioxidant assays used in this present work for the antiradical pharmacological evaluation of the target 1,3,4-oxadiazole derivatives 3n-z are the following ABTS test (2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) radical cation decolorization assay) and DPPH test (2,2-diphenyl-1-picrylhydrazyl free radical bleaching assay) which are two of the most common and famous assays used as a base for the further biological evaluation of the antioxidant activities of new organic compounds.
2.2.1. ABTS Test
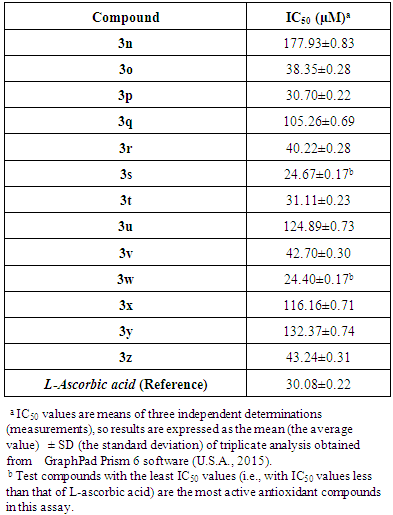
- ABTS test results (antioxidant IC50 values for the target compounds 3n-z) are summarized in Table 5 below. A comprehensive overview of the results in Table 5 reveals that the target compounds 3n-z displayed various degrees of free radical scavenging activity towards the ABTS radical, with decreasing activity in the following order: 3w > 3s > 3p > 3t > 3o > 3r > 3v > 3z > 3q > 3x > 3u > 3y > 3n. The most potent compounds are the two compounds 3w and 3s with in vitro antiradical effects (IC50 = 24.40 and 24.67 µM, respectively) higher than that of L-ascorbic acid (IC50 = 30.08 µM), while other compounds demonstrated either close and comparable activity to that of L-ascorbic acid (such as compounds 3p and 3t which have IC50 = 30.70 and 31.11 µM, respectively), or lower and weaker activity than that of L-ascorbic acid (such as compounds 3q, 3x, 3u, 3y, and 3n which have IC50 = 105.26, 116.16, 124.89, 132.37, and 177.93 µM, respectively).Compounds 3w and 3s are the most effective anti-ABTS·+ 1,3,4-oxadiazoles among all the thirteen derivatives due to the presence of relatively higher but balanced number of OH groups in their structures (in addition to the presence of complete strong conjugation and resonance in case of compound 3w), which strongly increases the in vitro antioxidant capacities of these two new compounds (as a result of the extensive hydrogen bonds formation and the higher aqueous solubility). On the other hand, compound 3n has relatively the lowest in vitro antioxidant activity among the thirteen 1,3,4-oxadiazole compounds possibly because its
 group is a very long straight aliphatic lipophilic chain as it consists of fifteen C atoms (i.e., not a small aliphatic structure) and this extremely inhibits the electron-donating effect of
group is a very long straight aliphatic lipophilic chain as it consists of fifteen C atoms (i.e., not a small aliphatic structure) and this extremely inhibits the electron-donating effect of  group on the oxadiazole ring (in vitro not in vivo) which, in turn, drastically decreases the free radical scavenging activity of the compound.
group on the oxadiazole ring (in vitro not in vivo) which, in turn, drastically decreases the free radical scavenging activity of the compound.
|
2.2.2. DPPH Test
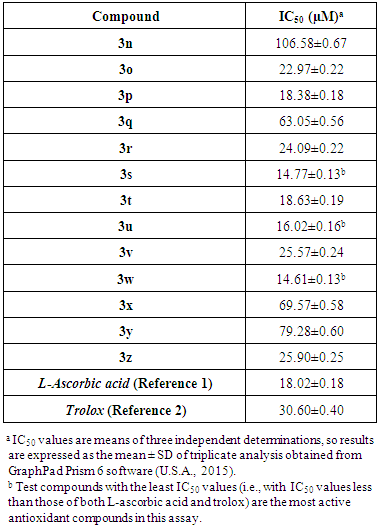
- DPPH test results (antioxidant IC50 values for the target compounds 3n-z) are summarized in Table 6. A complete overview of the results in Table 6 reveals that, exactly as with the ABTS radical, the target compounds 3n-z displayed various degrees of free radical scavenging activity towards the DPPH radical, with decreasing activity in the following order: 3w > 3s > 3u > 3p > 3t > 3o > 3r > 3v > 3z > 3q > 3x > 3y > 3n. The most potent compounds are the three compounds 3w, 3s, and 3u with in vitro antiradical effects (IC50 = 14.61, 14.77, and 16.02 µM, respectively) higher than that of both L-ascorbic acid and trolox (IC50 = 18.02 and 30.60 µM, respectively), while other compounds demonstrated either close and comparable activity to that of L-ascorbic acid with higher activity than that of trolox (such as compounds 3p and 3t which have IC50 = 18.38 and 18.63 µM, respectively), or lower and weaker activity than that of both L-ascorbic acid and trolox (only compounds 3q, 3x, 3y, and 3n which have IC50 = 63.05, 69.57, 79.28, and 106.58 µM, respectively). It was generally noted that most of the compounds 3n-z have stronger antioxidant activities than the potent antioxidant trolox.These results prove that compounds 3n-z are obviously good-excellent antioxidants (i.e., effective anti-DPPH· compounds). Being very close and relatively similar, the differences in values in this assay can be explained by and attributed to the same effects of structural modifications (i.e., differences) that were previously mentioned under ABTS test. The results of this DPPH assay are very closely related to those of ABTS assay (with the only exception, compound 3u, which has an excellent antiradical activity against DPPH· free radical, as its IC50 value equals about 16 µM, unlike its weak efficiency against the ABTS·+ radical cation) indicating that these new 1,3,4-oxadiazole derivatives behave with the same relative manner and efficiency against both radicals (DPPH· and ABTS·+), i.e., these new 1,3,4-oxadiazole derivatives react with and scavenge both types of radicals in closely efficient ways and this makes them having a wide spectrum of antioxidant activities against the different types of free radicals. An important observation is that the IC50 values of all the compounds 3n-z in DPPH assay are smaller than the corresponding ones of the same compounds in ABTS assay suggesting that these compounds are more potent scavengers of free radicals of the DPPH· type than those of the ABTS·+ type.
|
2.3. SAR (structure-antioxidant activity relationship)
- On correlating the modifications of the chemical structure (substituent
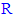 change) of the new compounds of the 5-substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles (i.e., along the new series of compounds 3n-z) with their in vitro antioxidant biological activity (in both ABTS and DPPH assays), it has been observed that:● Simple short-chain aliphatic-
change) of the new compounds of the 5-substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles (i.e., along the new series of compounds 3n-z) with their in vitro antioxidant biological activity (in both ABTS and DPPH assays), it has been observed that:● Simple short-chain aliphatic-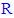 5-substituted-2-(3,4, 5-trihydroxyphenyl)-1,3,4-oxadiazole compounds are generally more active as antioxidants than large complicated aromatic-
5-substituted-2-(3,4, 5-trihydroxyphenyl)-1,3,4-oxadiazole compounds are generally more active as antioxidants than large complicated aromatic-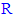 ones (supposing that there are not any additional moieties that affect the overall antioxidant activity).● As the length of the aliphatic straight chain (if present) at position 5 of the 1,3,4-oxadiazole compounds increases, the antioxidant activity of these compounds gradually decreases until it reaches certain limit (e.g., fifteen carbon atoms or more) above which, the compounds become much less active than those have short aliphatic straight chains (one to three carbon atoms) and also than aromatic-
ones (supposing that there are not any additional moieties that affect the overall antioxidant activity).● As the length of the aliphatic straight chain (if present) at position 5 of the 1,3,4-oxadiazole compounds increases, the antioxidant activity of these compounds gradually decreases until it reaches certain limit (e.g., fifteen carbon atoms or more) above which, the compounds become much less active than those have short aliphatic straight chains (one to three carbon atoms) and also than aromatic-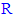 compounds (i.e., their antioxidant activities are relatively very weak), on a condition that there are not any additional moieties on the aliphatic chain that impart and add any antioxidant activity.● Aromatic-
compounds (i.e., their antioxidant activities are relatively very weak), on a condition that there are not any additional moieties on the aliphatic chain that impart and add any antioxidant activity.● Aromatic- 5-substituted-2-(3,4,5-trihydroxyphenyl)-1, 3,4-oxadiazoles having complete resonating system (uninterrupted resonance effect) are generally more active as antioxidants than those having incomplete interrupted one.● As the number of halogens (e.g., Cl and Br substituents) attached to the aliphatic side chain which is present at position 5 of the bioactive oxadiazole ring scaffold in aliphatic-
5-substituted-2-(3,4,5-trihydroxyphenyl)-1, 3,4-oxadiazoles having complete resonating system (uninterrupted resonance effect) are generally more active as antioxidants than those having incomplete interrupted one.● As the number of halogens (e.g., Cl and Br substituents) attached to the aliphatic side chain which is present at position 5 of the bioactive oxadiazole ring scaffold in aliphatic- 5-substituted-2-(3,4,5-trihydroxyphenyl)-1, 3,4-oxadiazole compounds increases, the antioxidant activity of these compounds decreases in a relative way.● Compounds that have considerable number of 1,3, 4-oxadiazole rings and 3,4,5-trihydroxyphenyl groups are generally expected to be very potent antioxidant compounds and to have much more antioxidant activities than those have only one 1,3,4-oxadiazole ring and one 3,4,5-trihydroxyphenyl group.●5-Substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles that have balanced lipophilic/hydrophilic properties are much more active in vitro as antioxidants than extremely lipophilic ones.
5-substituted-2-(3,4,5-trihydroxyphenyl)-1, 3,4-oxadiazole compounds increases, the antioxidant activity of these compounds decreases in a relative way.● Compounds that have considerable number of 1,3, 4-oxadiazole rings and 3,4,5-trihydroxyphenyl groups are generally expected to be very potent antioxidant compounds and to have much more antioxidant activities than those have only one 1,3,4-oxadiazole ring and one 3,4,5-trihydroxyphenyl group.●5-Substituted-2-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazoles that have balanced lipophilic/hydrophilic properties are much more active in vitro as antioxidants than extremely lipophilic ones.3. Conclusions
- On the basis of ‘‘the combination principles’’, we have designed and synthesized, in very good to excellent yields, a novel series of 5-substituted-2-(3,4,5-trihydroxyphenyl)-1, 3,4-oxadiazole compounds (compounds 3n-z) in which a bioactive aromatic 1,3,4-oxadiazole ring is directly linked with an antioxidant 3,4,5-trihydroxyphenyl moiety and an aiding substituent at the two carbons of the ring, the produced 2,5-disubstituted-1,3,4-oxadiazoles were characterized by most different spectral and elemental analytical methods. The synthesized compounds showed a wide range of potentially promising antioxidant activities. Depending on their pharmacological scavenging effects against the tested radicals in vitro, these target compounds can be categorized relative to L-ascorbic acid and trolox into three distinguished classes of antioxidants, as they can be classified into either very potent and excellent antioxidants (group I, compounds 3s,u,w), moderately potent and good antioxidants (group II, compounds 3o,p,r,t,v,z), or relatively less potent and mild antioxidants (group III, compounds 3n,q,x,y). Compounds 3s,u,w exhibited interestingly very potent antioxidant activity and they could be very promising lead and parent compounds for the design and synthesis of new antioxidant compounds and medicines by further in vivo biological evaluation, structural modifications, deep investigations, and advanced clinical studies.
4. Experimental Work
4.1. Chemical Synthesis
4.1.1. General Data
- All reactions were performed with commercially available reagents. All chemicals (reagents and solvents) were of analytical grade, purchased from commercial suppliers, and were used as received without further purification (if needed, some solvents were dried by standard methods). Acidic alumina (aluminum oxide; acidic; Brockmann I; ~ 150 mesh; 58 Å CAMAG 506-C-I; surface area = 155 m2/g; pH = 6) was used as an efficient adsorbent for the MW reactions of the last step. MWI (microwave irradiation) for MW reactions was carried out in an unmodified domestic MW oven (Samsung type, model M1733N with T.D.S. (Triple Distribution System) property, and having a power level of 100-800 W) operated at 2.45 GHz. TLC was used to monitor the progress of all the reactions to reach their completion (reaction times) and to check the purity of the compounds synthesized, it was carried out on TLC silica gel 60 F254 plates (plates of aluminum sheets precoated with unmodified silica gel 60 F254 to a layer thickness of 0.20 mm, purchased from E. Merck, Merck Millipore Division or Merck Chemicals, Merck KGaA, Darmstadt, Germany) as the stationary phase using petroleum ether/ethyl acetate/absolute EtOH (6:3:2, v/v/v “v = volume”) mixture (for all the reactions except for monitoring the hydrazinolysis reaction for the synthesis of compound 2nz in this work as a mixture of 5% absolute MeOH in CH2Cl2 was used as the mobile phase for it) as the eluting solvent system and the chromatograms spots were visualized and observed under the used UV (ultraviolet) light at wavelengths of 254 (mainly) and 366 nm to detect the produced components. The pH of reaction mixtures solutions was measured (to be adjusted) by a portable waterproof pH/ORP meter with smart electrodes and log-on-demand (HI 98150, HANNA instruments, Hungary Kft., Hungary) and this was done mainly to get the neutral pH (about 8) in the neutralization step for each reaction mixture solution contained POCl3 as excess with the product. Evaporation and concentration was carried out by using rotavap (rotary evaporator) under reduced pressure (for the efficient and gentle removal of solvents from reaction mixtures). A lyophilizer (freeze dryer, model FD8-8T, SIM international, U.S.A.) was used for lyophilizing purpose. Melting points (○C) of all the synthesized compounds were measured and recorded in open glass capillaries using Fisher-Johns melting point apparatus and were uncorrected. IR spectra were recorded on Nicolet™ iS™ 10 Mid-Infrared (ThermoFisher Scientific) FT-IR spectrometer (υ in cm-1) using KBr (potassium bromide) disks at the Central Laboratory (Faculty of Pharmacy, Mansoura University, Mansoura, Egypt) (Abbreviations in IR characterization data: Str. = Strong (if not mentioned, this means that the peak is weak to medium in intensity); Bro. = Broad (if not mentioned, this means that the peak is sharp, not broad enough, or overlapped with other peaks); Aliph. = Aliphatic; Arom. = Aromatic). 1H-NMR spectra were recorded on Varian Gemini-300 spectrometer (Mercury-300BB "NMR300") at about 300 MHz using TMS (tetramethylsilane) as an internal standard at the Microanalytical Center (Faculty of Science, Cairo University, Cairo, Egypt) and their chemical shifts values (δ) were given in ppm downfield from TMS at a temperature of 30 °C using either CDCl3 (deuterated chloroform) or DMSO-d6 (deuterated dimethylsulfoxide) as a solvent (according to the solubility of each analyzed compound) (Abbreviations in 1H-NMR characterization data: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; dd = double doublet (a doublet of doublets); J = Coupling Constant (expressed in Hz); Aliph. = Aliphatic; Arom. = Aromatic; o = ortho; m = meta; p = para). 13C-NMR spectrum for only compound 2nz was recorded also on Varian Gemini-300 spectrometer (Mercury-300BB "NMR300") at about 75 MHz using TMS as an internal standard at the Microanalytical Center (Faculty of Science, Cairo University, Cairo, Egypt) and its chemical shifts values (δ) were given in ppm downfield from TMS at a temperature of 30 °C using DMSO-d6 as a solvent (Abbreviation in 13C-NMR characterization data: Arom. = Aromatic). MS analyses were performed on Shimadzu Qp-2010 Plus at 70 eV and results were represented by m/z (Rel. Int.), i.e., mass/charge (relative intensity), at the Microanalytical Center (Faculty of Science, Cairo University, Cairo, Egypt). Elemental analyses were performed at the Microanalytical Center (Faculty of Science, Cairo University, Cairo, Egypt) in order to determine C, H, and N contents of all the newly synthesized compounds and the previously synthesized compound 2nz (they, all, were in full agreement with the calculated theoretical values). Other abbreviations in synthetic procedures and characterization data below: abs. = absolute; R.T. = room temperature; EtOAc = ethyl acetate; M.P. = Melting point; Recryst. = Recrystallized; DEE = diethyl ether; v/v = volume per volume; Col. & App. = Color & Appearance; M.Wt. = Molecular Weight; Elem. Anal. = Elemental Analyses; DMF = dimethylformamide; dec. = with decomposition.
4.1.2. Conventional Synthesis of Ethyl 3,4,5-Trihydroxybenzoate (1nz)
- To a stirred solution of anhydrous gallic acid (0.1 mole, 17.012 g) in abs. EtOH (500 mL) in a round-bottomed flask, Conc. H2SO4 (5 mL) was added. The resulted reaction mixture was refluxed with stirring at 90-100 °C for 15 h. After reaction completion (i.e., no more H2O was being distilled off), the reaction mixture was cooled to R.T., then excess EtOH was completely evaporated (distilled off) on a water bath, and the reaction mixture was allowed to cool to R.T. again. The residue remained was taken up in pure distilled H2O in a separatory funnel and the flask was rinsed with a few mL of pure distilled H2O which were also poured into the separatory funnel. The formed aqueous solution was extracted by shaking it vigorously with EtOAc for three times (3 × 250 mL; the upper organic layer was carefully taken each time while the lower aqueous layer was finally rejected), the combined organic layers (extracts) were returned to a pure dry separatory funnel to be subsequently shaken with a strong solution of NaHCO3 (sodium bicarbonate) until all the free gallic acid was removed (i.e., until no more effervescence), then they were washed with distilled H2O and brine (a highly concentrated NaCl (sodium chloride) solution in H2O). The organic layer was taken and dried over anhydrous Na2SO4 (sodium sulfate) during filtration (directly into the pure dry round-bottomed flask of the rotavap), then the filtrate was concentrated by evaporation in rotavap under reduced pressure, left in air to be completely dry to give a crude white solid mass of the product 1nz which upon further purification by recrystallization from hexane, it gives the pure product 1nz (16.508 g, yield ≈ 83.3% (as reported [26,27])) which is a white amorphous powder with M.P. = 148-150 °C (as reported [26,27]).
4.1.3. Synthesis of 3,4,5-Trihydroxybenzohydrazide (2nz)
- 2nz was synthesized by the following two methods:
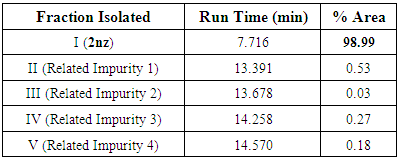
 A mixture of 1nz (0.1 mole, 19.817 g) and slight excess NH2NH2.H2O (0.11 mole; about 5.35 mL) was dissolved in abs. EtOH (about 300-350 mL, i.e., the least amount needed to make the reaction mixture a clear solution) in a round-bottomed flask, then the resulted reaction mixture was mixed gently and refluxed with stirring for 6 h. The progress of the reaction was monitored on TLC plates. After reaction completion, the reaction mixture was cooled to R.T., then excess EtOH was completely distilled off on a water bath, and the reaction mixture was allowed to cool to R.T. again. The reaction mixture was poured onto ice-cold water with stirring and the white mass or precipitate obtained was filtered, washed with distilled H2O several times, and dried under vacuum (or left in air to be completely dry) to give a crude pale white solid mass of the product 2nz which was further purified by HPLC analysis (2nz was successfully obtained with very excellent purity of about 99%; see details below) and recryst. twice from DEE and abs. MeOH (1:1, v/v) to give about 16.574 g (yield = 90.0% (as reported [37])) of the pure 2nz (pale white to buff fine powder) with M.P. = 294-296 °C (reported one = 290 °C [48]).
A mixture of 1nz (0.1 mole, 19.817 g) and slight excess NH2NH2.H2O (0.11 mole; about 5.35 mL) was dissolved in abs. EtOH (about 300-350 mL, i.e., the least amount needed to make the reaction mixture a clear solution) in a round-bottomed flask, then the resulted reaction mixture was mixed gently and refluxed with stirring for 6 h. The progress of the reaction was monitored on TLC plates. After reaction completion, the reaction mixture was cooled to R.T., then excess EtOH was completely distilled off on a water bath, and the reaction mixture was allowed to cool to R.T. again. The reaction mixture was poured onto ice-cold water with stirring and the white mass or precipitate obtained was filtered, washed with distilled H2O several times, and dried under vacuum (or left in air to be completely dry) to give a crude pale white solid mass of the product 2nz which was further purified by HPLC analysis (2nz was successfully obtained with very excellent purity of about 99%; see details below) and recryst. twice from DEE and abs. MeOH (1:1, v/v) to give about 16.574 g (yield = 90.0% (as reported [37])) of the pure 2nz (pale white to buff fine powder) with M.P. = 294-296 °C (reported one = 290 °C [48]). A mixture of gallic acid (0.01 mole, 1.7012 g) and slight excess NH2NH2.H2O (0.012 mole; about 0.584 mL) was taken in a 150-mL conical flask, the resulted paste of the reaction mixture was well mixed, then the flask was covered with aluminum foil and subjected to MWI intermittently at intervals of 30 s for 2 min (i.e., 4 intervals of 30 s) at a power level of 800 W. The progress of the reaction was monitored on TLC plates till it was over after the fourth interval. After reaction completion, the reaction mixture was cooled to -20 °C and then it was lyophilized at -50 °C. The white product obtained was washed with distilled H2O several times and dried under vacuum (or left in air to be completely dry) to give a crude pale white solid mass of the product 2nz which was further purified by HPLC analysis (2nz was successfully obtained with very excellent purity of about 99%; see details below) and recryst. twice from DEE and abs. MeOH (1:1, v/v) to give about 1.8084 g (yield = 98.2%) of the pure 2nz (pale white to buff fine powder) with M.P. = 294-296 °C (reported one = 290 °C [48]).● HPLC analysis for separation of 2nz and determination of its crude product purity:For these two purposes, the following HPLC analytical conditions were used: - Column Type: SYMMETRY SHIELD RP C18 (250 × 4.6 mm; 5 μm; equipped with Empower 2 software).- Column Temperature: Ambient.- Mobile Phase Constituents: A = MeOH; B = 0.1% TFA (trifluoroacetic acid) in H2O.- Diluent Constitution: ACN (acetonitrile) + TFA.- Injection Volume: 2 μL.- Flow Rate: 1 mL/min.- Run Time: 20 min.- Gradient Solvent System (Time (min)/A (%)): 0/0; 6/0; 10/70; 18/70; 19/0; 20/0.- Wavelengths for UV Detection: 220 & 272 nm.
A mixture of gallic acid (0.01 mole, 1.7012 g) and slight excess NH2NH2.H2O (0.012 mole; about 0.584 mL) was taken in a 150-mL conical flask, the resulted paste of the reaction mixture was well mixed, then the flask was covered with aluminum foil and subjected to MWI intermittently at intervals of 30 s for 2 min (i.e., 4 intervals of 30 s) at a power level of 800 W. The progress of the reaction was monitored on TLC plates till it was over after the fourth interval. After reaction completion, the reaction mixture was cooled to -20 °C and then it was lyophilized at -50 °C. The white product obtained was washed with distilled H2O several times and dried under vacuum (or left in air to be completely dry) to give a crude pale white solid mass of the product 2nz which was further purified by HPLC analysis (2nz was successfully obtained with very excellent purity of about 99%; see details below) and recryst. twice from DEE and abs. MeOH (1:1, v/v) to give about 1.8084 g (yield = 98.2%) of the pure 2nz (pale white to buff fine powder) with M.P. = 294-296 °C (reported one = 290 °C [48]).● HPLC analysis for separation of 2nz and determination of its crude product purity:For these two purposes, the following HPLC analytical conditions were used: - Column Type: SYMMETRY SHIELD RP C18 (250 × 4.6 mm; 5 μm; equipped with Empower 2 software).- Column Temperature: Ambient.- Mobile Phase Constituents: A = MeOH; B = 0.1% TFA (trifluoroacetic acid) in H2O.- Diluent Constitution: ACN (acetonitrile) + TFA.- Injection Volume: 2 μL.- Flow Rate: 1 mL/min.- Run Time: 20 min.- Gradient Solvent System (Time (min)/A (%)): 0/0; 6/0; 10/70; 18/70; 19/0; 20/0.- Wavelengths for UV Detection: 220 & 272 nm.
|
4.1.4. Synthesis of 5-(5-substituted-1,3,4-oxadiazol-2 -yl)benzene-1,2,3-triols (3n-z)
- The target compounds 3n-z were synthesized by the following two methods:
 An ice-cooled mixture of 2nz (0.01 mole, 1.8415 g if the carboxylic acid is monocarboxylic acid; 0.02 mole, 3.6830 g if the carboxylic acid is dicarboxylic acid; or 0.03 mole, 5.5245 g if the carboxylic acid is tricarboxylic acid) and the respective carboxylic acid (0.01 mole; see Table 3) was dissolved in dry POCl3 (5 mL if the carboxylic acid is monocarboxylic acid, 10 mL if the carboxylic acid is dicarboxylic acid, or 15 mL if the carboxylic acid is tricarboxylic acid; by dropwise addition of POCl3 to the mixture) and the resulted solution was gently heated under reflux (i.e., at about 105-110 °C) with constant magnetic stirring for 3-9 h (see Table 3). The reaction was monitored and followed up by using TLC plates. When the reaction was over as indicated by TLC, the reaction mixture solution was concentrated in rotavap under reduced pressure, cooled to R.T., and then gradually and carefully poured onto crushed ice with stirring. The least amount required of finely powdered K2CO3 (potassium carbonate) and the required amount of solid KOH (potassium hydroxide) were added with stirring to the mixture solution till the pH of the solution was raised to 8 (it was measured by using pHmeter) to remove the excess of POCl3. The mixture solution was allowed to stand overnight till the solid was separated and completely precipitated. The separated crude solid was filtered, washed thoroughly with cold distilled H2O, dried, and purified by recrystallization from an appropriate solvent or mixture of solvents (see for each compound below) to give the pure product 3 as shown below in details.
An ice-cooled mixture of 2nz (0.01 mole, 1.8415 g if the carboxylic acid is monocarboxylic acid; 0.02 mole, 3.6830 g if the carboxylic acid is dicarboxylic acid; or 0.03 mole, 5.5245 g if the carboxylic acid is tricarboxylic acid) and the respective carboxylic acid (0.01 mole; see Table 3) was dissolved in dry POCl3 (5 mL if the carboxylic acid is monocarboxylic acid, 10 mL if the carboxylic acid is dicarboxylic acid, or 15 mL if the carboxylic acid is tricarboxylic acid; by dropwise addition of POCl3 to the mixture) and the resulted solution was gently heated under reflux (i.e., at about 105-110 °C) with constant magnetic stirring for 3-9 h (see Table 3). The reaction was monitored and followed up by using TLC plates. When the reaction was over as indicated by TLC, the reaction mixture solution was concentrated in rotavap under reduced pressure, cooled to R.T., and then gradually and carefully poured onto crushed ice with stirring. The least amount required of finely powdered K2CO3 (potassium carbonate) and the required amount of solid KOH (potassium hydroxide) were added with stirring to the mixture solution till the pH of the solution was raised to 8 (it was measured by using pHmeter) to remove the excess of POCl3. The mixture solution was allowed to stand overnight till the solid was separated and completely precipitated. The separated crude solid was filtered, washed thoroughly with cold distilled H2O, dried, and purified by recrystallization from an appropriate solvent or mixture of solvents (see for each compound below) to give the pure product 3 as shown below in details. An ice-cooled mixture of 2nz (0.01 mole, 1.8415 g if the carboxylic acid is monocarboxylic acid; 0.02 mole, 3.6830 g if the carboxylic acid is dicarboxylic acid; or 0.03 mole, 5.5245 g if the carboxylic acid is tricarboxylic acid) and the respective carboxylic acid (0.01 mole; see Table 3) was dissolved in dry POCl3 (5 mL if the carboxylic acid is monocarboxylic acid, 10 mL if the carboxylic acid is dicarboxylic acid, or 15 mL if the carboxylic acid is tricarboxylic acid; by dropwise addition of POCl3 to the mixture); acidic alumina (5 g if the carboxylic acid is monocarboxylic acid, 10 g if the carboxylic acid is dicarboxylic acid, or 15 g if the carboxylic acid is tricarboxylic acid) was added to the above-resulted solution at R.T.; and the resulted paste of reaction mixture was well mixed, adsorbed, dried, kept inside the alumina bath, covered with aluminum foil, and subjected to intermittent MWI at intervals of 30 s for 4-10 min at a power level of 300-800 W (see Table 3). The reaction was monitored and followed up by using TLC plates till it was over. After cooling the reaction mixture to R.T., a suitable amount of anhydrous CH2Cl2 was added to this mixture to efficiently dissolve the acidic alumina; the CH2Cl2 layer was evaporated in rotavap under reduced pressure to completely remove the acidic alumina from the reaction mixture; the remaining crude paste was cooled to R.T. and then gradually poured with care onto crushed ice with stirring. The least amount required of finely powdered K2CO3 and the required amount of solid KOH were added with stirring to the mixture solution till the pH of the solution was raised to 8 (it was measured by using pHmeter) to remove the excess of POCl3. The mixture solution was allowed to stand overnight till the solid was separated and completely settled down. The separated crude solid was filtered, washed thoroughly with cold distilled H2O, dried, and purified by recrystallization from an appropriate solvent(s) (see for each compound below) to give the pure product 3 as shown below in details.● 5-(5-Pentadecyl-1,3,4-oxadiazol-2-yl)benzene-1,2, 3-triol (3n): Recryst. from benzene; Col. & App.: pale buff fine powder; Yield: 94.0% (Conv.), 99.2% (MW); M.P.: 114 °C; IR (υ in cm-1): Str. & Bro. 3452 (O-H), Str. 3129 (C-H, Arom.), Str. 2909 (C-H, Aliph.), 1660 (C=N), 1532 & 1519 & 1377 (C=C, Arom.), Str. 1263 & Str. 1137 (C-O), Str. 1063 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 0.843-0.932 (t, J = 8.0 Hz, 3H, Terminal CH3), 1.259-1.679 (m, 26H, All Other 13 CH2), 2.495-2.513 (t, J = 7.1 Hz, 2H, α-CH2 to Oxadiazole Ring), 5.391 (s, 3H, 3 Arom. OH), 6.785-7.313 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 404.54): 405.10 (8.90), 388.10 (8.28), 280.10 (10.12), 125.10 (17.33), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C23H36N2O4): Calculated (Found): C: 68.29 (68.18), H: 8.97 (8.93), N: 6.92 (6.93).● 5-[5-(1-Hydroxyethyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3o): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: greyish green fine powder; Yield: 92.0% (Conv.), 98.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3452 (O-H), Str. 3094 (C-H, Arom.), Str. 2922 (C-H, Aliph.), Str. 1579 (C=N), 1505 & 1466 & 1401 & Str. 1371 (C=C, Arom.), Str. 1263 (C-O), Str. 1048 (N-N); 1H-NMR (CDCl3, δ in ppm): 1.267-1.582 (d, J = 6.8 Hz, 3H, CH3), 3.653 (s, 1H, Aliph. OH), 4.676-4.686 (q, J = 6.8 Hz, 1H, CH), 5.373 (s, 3H, 3 Arom. OH), 7.272 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 238.20): 238.25 (1.70), 221.20 (0.99), 125.15 (13.89), 113.15 (15.33), 69.05 (35.66), 57.05 (100.00); Elem. Anal. (%, for C10H10N2O5): Calculated (Found): C: 50.42 (50.46), H: 4.23 (4.24), N: 11.76 (11.73).● 5-[5-(Dichloromethyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3p): Recryst. from EtOAc; Col. & App.: dark brownish green fine powder; Yield: 90.3% (Conv.), 97.3% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3372 (O-H), Str. 2922 (C-H, Arom.), Str. 2850 (C-H, Aliph.), Str. 1678 (C=N), Str. 1578 & Str. 1509 & Str. 1465 & Str. 1404 & Str. 1373 (C=C, Arom.), 1242 & Str. 1199 (C-O), Str. 1051 (N-N), 766 & 750 (C-Cl); 1H-NMR (CDCl3, δ in ppm): 5.548 (s, 3H, 3 Arom. OH), 7.272 (2 Overlapped s, 3H, 2 Benzene-H & CH); MS (m/z (Rel. Int. in %), M.Wt. = 277.06): 277.25 (2.01), 152.15 (2.02), 125.15 (17.89), 83.10 (33.62), 69.05 (39.41), 57.05 (100.00); Elem. Anal. (%, for C9H6Cl2N2O4): Calculated (Found): C: 39.02 (39.01), H: 2.18 (2.18), N: 10.11 (10.10).● 5-[5-(Trichloromethyl)-1,3,4-oxadiazol-2-yl]benzene-1,2,3-triol (3q): Recryst. from EtOAc; Col. & App.: pale white crystalline powder; Yield: 89.1% (Conv.), 96.4% (MW); M.P.: 56-58 °C; IR (υ in cm-1): Str. & Bro. 3466 (O-H), Str. 2922 (C-H, Arom.), Str. 1667 & Str. 1633 (C=N), 1557 & 1538 & 1514 & 1504 & 1463 (C=C, Arom.), Str. 1199 (C-O), Str. 1077 (N-N), Str. 868 (C-Cl); 1H-NMR (CDCl3, δ in ppm): 5.359 (s, 3H, 3 Arom. OH), 7.270 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 311.51): 311.10 (33.20), 294.10 (23.08), 187.10 (8.91), 125.15 (2.02), 69.05 (28.34), 59.00 (100.00); Elem. Anal. (%, for C9H5Cl3N2O4): Calculated (Found): C: 34.70 (34.77), H: 1.62 (1.61), N: 8.99 (8.95).● (S)-5-[5-(1-Amino-2-phenylethyl)-1,3,4-oxadiazol-2- yl]benzene-1,2,3-triol (3r): Recryst. from DEE/abs. EtOH (3:1, v/v); Col. & App.: pale white minute crystalline plates; Yield: 83.5% (Conv.), 97.9% (MW); M.P.: 256-258 °C (dec.); IR (υ in cm-1): Str. & Bro. 3427 (O-H), Str. 3388 & Str. 3299 (2 N-H, i.e., NH2), Str. 2922 (C-H, Arom.), Str. 2850 (C-H, Aliph.), 1663 (C=N), Str. 1607 & 1548 & Str. 1500 & 1467 & 1367 (C=C, Arom.), 1286 (C-N, Aliph.), 1211 (C-O), Str. 1078 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 3.073-3.087 & 3.097-3.112 (2 dd, J = -12.4 Hz & 7.0 Hz, 2 Diastereotopic H, CH2), 4.351 (t, J = 7.0 Hz, 1H, CH), 5.113 (s, 2H, Aliph. NH2), 5.364 (s, 3H, 3 Arom. OH), 6.787 (s, 2H, 2 Benzene-H), 7.275-7.327 (m, 5H, 1 p- & 2 o- & 2 m-Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 313.31): 313.10 (2.44), 222.00 (26.99), 153.00 (100.00), 125.05 (38.21), 91.05 (63.36), 69.00 (2.73); Elem. Anal. (%, for C16H15N3O4): Calculated (Found): C: 61.34 (61.39), H: 4.83 (4.80), N: 13.41 (13.41).● 5,5'-[5,5'-Methylenebis(1,3,4-oxadiazole-5,2-diyl)]di- benzene-1,2,3-triol (3s): Recryst. from DMF/abs. EtOH (2:1, v/v); Col. & App.: whitish buff crystalline powder; Yield: 84.0% (Conv.), 95.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3424 (O-H), 2922 (C-H, Arom.), 2850 (C-H, Aliph.), 1659 & 1651 (C=N), 1615 & 1548 & 1538 & 1504 & 1469 & 1455 (C=C, Arom.), 1285 & 1208 (C-O), Str. 1083 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 3.366-4.276 (s, 2H, CH2), 5.368 (s, 6H, 6 Arom. OH), 6.777-6.781 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 400.30): 400.10 (18.29), 109.10 (19.54), 83.10 (31.08), 69.10 (42.27), 57.05 (100.00), 55.00 (51.87); Elem. Anal. (%, for C17H12N4O8): Calculated (Found): C: 51.01 (51.09), H: 3.02 (3.03), N: 14.00 (14.04).● 5,5'-{5,5'-[(1R,2R)-1,2-Dihydroxyethane-1,2-diyl]bis(1,3, 4-oxadiazole-5,2-diyl)}dibenzene-1,2,3-triol (3t): Recryst. from DMF/abs. EtOH (2:1, v/v); Col. & App.: brownish orange fine powder; Yield: 75.0% (Conv.), 97.0% (MW); M.P.: 198-200 °C; IR (υ in cm-1): Str. & Bro. 3416 (O-H), Str. 2972 (C-H, Arom.), Str. 2850 (C-H, Aliph.), Str. 1682 & Str. 1616 (C=N), 1551 & 1538 & 1531 & 1515 & 1449 & 1398 (C=C, Arom.), Str. 1240 & 1186 (C-O), 1099 & Str. 1030 (N-N); 1H-NMR (CDCl3, δ in ppm): 3.655 (s, 2H, 2 Aliph. OH), 4.918-4.971 (d, J = 7 Hz, 2H, 2 CH), 5.375 (s, 6H, 6 Arom. OH), 7.259-7.272 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 446.32): 447.00 (9.91), 446.00 (7.73), 125.00 (27.95), 79.95 (100.00), 69.00 (42.61), 57.05 (73.13); Elem. Anal. (%, for C18H14N4O10): Calculated (Found): C: 48.44 (48.48), H: 3.16 (3.14), N: 12.55 (12.53).● 1,2,3-Tris[5-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazol-2- yl]propan-2-ol (3u): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brown crystalline powder; Yield: 90.0% (Conv.), 99.0% (MW); M.P.: 99-100 °C (dec.); IR (υ in cm-1): Str. & Bro. 3460 (O-H), 2917 (C-H, Arom.), 2850 (C-H, Aliph.), Str. 1637 (C=N), 1489 & 1389 (C=C, Arom.), 1262 (C-O), Str. 1071 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 2.495-2.507 (s, 4H, 2 CH2), 3.338 (s, 1H, Aliph. OH), 5.373 (s, 9H, 9 Arom. OH), 7.270 (s, 6H, 6 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 636.48): 636.00 (4.35), 443.00 (3.38), 125.00 (5.69), 69.00 (22.54), 63.95 (100.00), 57.05 (37.95); Elem. Anal. (%, for C27H20N6O13): Calculated (Found): C: 50.95 (50.96), H: 3.17 (3.16), N: 13.20 (13.21).● (E)-5-(5-Styryl-1,3,4-oxadiazol-2-yl)benzene-1,2,3-triol (3v): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brownish green crystalline powder; Yield: 73.0% (Conv.), 97.5% (MW); M.P.: 299-300 °C (dec.); IR (υ in cm-1): Str. & Bro. 3445 (O-H), Str. 3080 & Str. 3055 (=C-H, Alkene), Str. 2918 (C-H, Arom.), Str. 1641 (C=C, Alkene), 1578 (C=N), 1556 & Str. 1514 & Str. 1450 & Str. 1387 (C=C, Arom.), 1257 & 1230 & 1203 (C-O), Str. 1072 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 5.351 (s, 3H, 3 Arom. OH), 6.787 (s, 2H, 2 Benzene-H), 6.955 & 7.029 (2 d, J = 15.1 Hz, 2H, trans HC=CH), 7.282-7.327 (m, 5H, 1 p- & 2 m- & 2 o-Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 296.28): 296.10 (0.83), 153.00 (100.00), 125.05 (38.21), 103.05 (6.93), 77.00 (7.06), 69.00 (2.73); Elem. Anal. (%, for C16H12N2O4): Calculated (Found): C: 64.86 (64.80), H: 4.08 (4.09), N: 9.46 (9.49).● (E)-5,5'-[5,5'-(Ethene-1,2-diyl)bis(1,3,4-oxadiazole-5, 2-diyl)]dibenzene-1,2,3-triol (3w): Recryst. from abs. MeOH; Col. & App.: light brown fine powder; Yield: 93.2% (Conv.), 98.8% (MW); M.P.: 288-290 °C (dec.); IR (υ in cm-1): Str. & Bro. 3425 (O-H), Str. 3077 & Str. 3054 (=C-H, Alkene), Str. 2926 (C-H, Arom.), Str. 1602 (C=C, Alkene), 1546 (C=N), 1505 & 1467 & 1413 & 1373 (C=C, Arom.), 1286 & 1206 (C-O), Str. 1078 & Str. 1057 (N-N); 1H-NMR (CDCl3, δ in ppm): 5.362 (s, 6H, 6 Arom. OH), 6.776 (s, 4H, 4 Benzene-H), 6.788 (d, J = 15.1 Hz, 2H, trans HC=CH); MS (m/z (Rel. Int. in %), M.Wt. = 412.31): 412.10 (19.36), 111.10 (22.38), 95.10 (20.60), 71.10 (53.82), 69.10 (42.27), 57.05 (100.00); Elem. Anal. (%, for C18H12N4O8): Calculated (Found): C: 52.43 (52.47), H: 2.93 (2.92), N: 13.59 (13.57).● 5-[5-(4-Bromophenyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3x): Recryst. from abs. EtOH; Col. & App.: buff crystalline powder; Yield: 82.8% (Conv.), 96.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3476 (O-H), Str. 3066 (C-H, Arom.), 1637 (C=N), 1551 & 1517 & 1499 & 1374 (C=C, Arom.), 1189 (C-O), Str. 1079 (N-N), Str. 530 (C-Br); 1H-NMR (DMSO-d6, δ in ppm): 5.382 (s, 3H, 3 Arom. OH), 6.785-6.882 (s, 2H, 2 Benzene-H), 7.305-7.313 (m, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 349.14): 349.10 (8.90), 193.10 (10.12), 157.10 (10.74), 125.10 (17.33), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C14H9BrN2O4): Calculated (Found): C: 48.16 (48.12), H: 2.60 (2.63), N: 8.02 (8.06).● 5,5'-(1,3,4-Oxadiazole-2,5-diyl)dibenzene-1,2,3-triol (3y): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brown crystalline powder; Yield: 88.0% (Conv.), 98.2% (MW); M.P.: 82-84 °C; IR (υ in cm-1): Str. & Bro. 3466 (O-H), Str. 3090 (C-H, Arom.), 1647 (C=N), 1553 & 1533 & 1468 & 1380 (C=C, Arom.), 1199 (C-O), Str. 1071 (N-N); 1H-NMR (CDCl3, δ in ppm): 5.333 (s, 6H, 6 Arom. OH), 6.736 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 318.24): 318.10 (2.44), 193.00 (0.90), 153.00 (100.00), 125.05 (38.21), 69.00 (2.73), 57.00 (2.59); Elem. Anal. (%, for C14H10N2O7): Calculated (Found): C: 52.84 (52.91), H: 3.17 (3.15), N: 8.80 (8.76).● 5-[5-(7-Chloro-4-hydroxyquinolin-3-yl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3z): Recryst. from hexane; Col. & App.: brownish buff fine powder; Yield: 86.4% (Conv.), 95.5% (MW); M.P.: 60-61 °C (dec.); IR (υ in cm-1): Str. & Bro. 3467 (O-H), Str. 3100 (C-H, Arom.), Str. 1620 (C=N), 1533 & 1483 & 1457 & 1378 (C=C, Arom.), Str. 1081 (C-O), Str. 983 (N-N), 869 (C-Cl); 1H-NMR (DMSO-d6, δ in ppm): 5.337 (s, 4H, 4 Arom. OH), 6.716 (s, 2H, 2 Benzene-H), 7.707 (dd, J = 7.5 Hz & 1.5 Hz, 1H, Quinoline-H-7), 7.949 (d, J = 1.5 Hz, 1H, Quinoline-H-9), 8.406 & 8.428 (d & s, JH-6 = 7.5 Hz, 2H, Quinoline-H-6,2); MS (m/z (Rel. Int. in %), M.Wt. = 371.73): 371.10 (9.51), 193.10 (10.12), 144.10 (9.20), 129.10 (28.07), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C17H10ClN3O5): Calculated (Found): C: 54.93 (54.91), H: 2.71 (2.72), N: 11.30 (11.33).
An ice-cooled mixture of 2nz (0.01 mole, 1.8415 g if the carboxylic acid is monocarboxylic acid; 0.02 mole, 3.6830 g if the carboxylic acid is dicarboxylic acid; or 0.03 mole, 5.5245 g if the carboxylic acid is tricarboxylic acid) and the respective carboxylic acid (0.01 mole; see Table 3) was dissolved in dry POCl3 (5 mL if the carboxylic acid is monocarboxylic acid, 10 mL if the carboxylic acid is dicarboxylic acid, or 15 mL if the carboxylic acid is tricarboxylic acid; by dropwise addition of POCl3 to the mixture); acidic alumina (5 g if the carboxylic acid is monocarboxylic acid, 10 g if the carboxylic acid is dicarboxylic acid, or 15 g if the carboxylic acid is tricarboxylic acid) was added to the above-resulted solution at R.T.; and the resulted paste of reaction mixture was well mixed, adsorbed, dried, kept inside the alumina bath, covered with aluminum foil, and subjected to intermittent MWI at intervals of 30 s for 4-10 min at a power level of 300-800 W (see Table 3). The reaction was monitored and followed up by using TLC plates till it was over. After cooling the reaction mixture to R.T., a suitable amount of anhydrous CH2Cl2 was added to this mixture to efficiently dissolve the acidic alumina; the CH2Cl2 layer was evaporated in rotavap under reduced pressure to completely remove the acidic alumina from the reaction mixture; the remaining crude paste was cooled to R.T. and then gradually poured with care onto crushed ice with stirring. The least amount required of finely powdered K2CO3 and the required amount of solid KOH were added with stirring to the mixture solution till the pH of the solution was raised to 8 (it was measured by using pHmeter) to remove the excess of POCl3. The mixture solution was allowed to stand overnight till the solid was separated and completely settled down. The separated crude solid was filtered, washed thoroughly with cold distilled H2O, dried, and purified by recrystallization from an appropriate solvent(s) (see for each compound below) to give the pure product 3 as shown below in details.● 5-(5-Pentadecyl-1,3,4-oxadiazol-2-yl)benzene-1,2, 3-triol (3n): Recryst. from benzene; Col. & App.: pale buff fine powder; Yield: 94.0% (Conv.), 99.2% (MW); M.P.: 114 °C; IR (υ in cm-1): Str. & Bro. 3452 (O-H), Str. 3129 (C-H, Arom.), Str. 2909 (C-H, Aliph.), 1660 (C=N), 1532 & 1519 & 1377 (C=C, Arom.), Str. 1263 & Str. 1137 (C-O), Str. 1063 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 0.843-0.932 (t, J = 8.0 Hz, 3H, Terminal CH3), 1.259-1.679 (m, 26H, All Other 13 CH2), 2.495-2.513 (t, J = 7.1 Hz, 2H, α-CH2 to Oxadiazole Ring), 5.391 (s, 3H, 3 Arom. OH), 6.785-7.313 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 404.54): 405.10 (8.90), 388.10 (8.28), 280.10 (10.12), 125.10 (17.33), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C23H36N2O4): Calculated (Found): C: 68.29 (68.18), H: 8.97 (8.93), N: 6.92 (6.93).● 5-[5-(1-Hydroxyethyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3o): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: greyish green fine powder; Yield: 92.0% (Conv.), 98.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3452 (O-H), Str. 3094 (C-H, Arom.), Str. 2922 (C-H, Aliph.), Str. 1579 (C=N), 1505 & 1466 & 1401 & Str. 1371 (C=C, Arom.), Str. 1263 (C-O), Str. 1048 (N-N); 1H-NMR (CDCl3, δ in ppm): 1.267-1.582 (d, J = 6.8 Hz, 3H, CH3), 3.653 (s, 1H, Aliph. OH), 4.676-4.686 (q, J = 6.8 Hz, 1H, CH), 5.373 (s, 3H, 3 Arom. OH), 7.272 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 238.20): 238.25 (1.70), 221.20 (0.99), 125.15 (13.89), 113.15 (15.33), 69.05 (35.66), 57.05 (100.00); Elem. Anal. (%, for C10H10N2O5): Calculated (Found): C: 50.42 (50.46), H: 4.23 (4.24), N: 11.76 (11.73).● 5-[5-(Dichloromethyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3p): Recryst. from EtOAc; Col. & App.: dark brownish green fine powder; Yield: 90.3% (Conv.), 97.3% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3372 (O-H), Str. 2922 (C-H, Arom.), Str. 2850 (C-H, Aliph.), Str. 1678 (C=N), Str. 1578 & Str. 1509 & Str. 1465 & Str. 1404 & Str. 1373 (C=C, Arom.), 1242 & Str. 1199 (C-O), Str. 1051 (N-N), 766 & 750 (C-Cl); 1H-NMR (CDCl3, δ in ppm): 5.548 (s, 3H, 3 Arom. OH), 7.272 (2 Overlapped s, 3H, 2 Benzene-H & CH); MS (m/z (Rel. Int. in %), M.Wt. = 277.06): 277.25 (2.01), 152.15 (2.02), 125.15 (17.89), 83.10 (33.62), 69.05 (39.41), 57.05 (100.00); Elem. Anal. (%, for C9H6Cl2N2O4): Calculated (Found): C: 39.02 (39.01), H: 2.18 (2.18), N: 10.11 (10.10).● 5-[5-(Trichloromethyl)-1,3,4-oxadiazol-2-yl]benzene-1,2,3-triol (3q): Recryst. from EtOAc; Col. & App.: pale white crystalline powder; Yield: 89.1% (Conv.), 96.4% (MW); M.P.: 56-58 °C; IR (υ in cm-1): Str. & Bro. 3466 (O-H), Str. 2922 (C-H, Arom.), Str. 1667 & Str. 1633 (C=N), 1557 & 1538 & 1514 & 1504 & 1463 (C=C, Arom.), Str. 1199 (C-O), Str. 1077 (N-N), Str. 868 (C-Cl); 1H-NMR (CDCl3, δ in ppm): 5.359 (s, 3H, 3 Arom. OH), 7.270 (s, 2H, 2 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 311.51): 311.10 (33.20), 294.10 (23.08), 187.10 (8.91), 125.15 (2.02), 69.05 (28.34), 59.00 (100.00); Elem. Anal. (%, for C9H5Cl3N2O4): Calculated (Found): C: 34.70 (34.77), H: 1.62 (1.61), N: 8.99 (8.95).● (S)-5-[5-(1-Amino-2-phenylethyl)-1,3,4-oxadiazol-2- yl]benzene-1,2,3-triol (3r): Recryst. from DEE/abs. EtOH (3:1, v/v); Col. & App.: pale white minute crystalline plates; Yield: 83.5% (Conv.), 97.9% (MW); M.P.: 256-258 °C (dec.); IR (υ in cm-1): Str. & Bro. 3427 (O-H), Str. 3388 & Str. 3299 (2 N-H, i.e., NH2), Str. 2922 (C-H, Arom.), Str. 2850 (C-H, Aliph.), 1663 (C=N), Str. 1607 & 1548 & Str. 1500 & 1467 & 1367 (C=C, Arom.), 1286 (C-N, Aliph.), 1211 (C-O), Str. 1078 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 3.073-3.087 & 3.097-3.112 (2 dd, J = -12.4 Hz & 7.0 Hz, 2 Diastereotopic H, CH2), 4.351 (t, J = 7.0 Hz, 1H, CH), 5.113 (s, 2H, Aliph. NH2), 5.364 (s, 3H, 3 Arom. OH), 6.787 (s, 2H, 2 Benzene-H), 7.275-7.327 (m, 5H, 1 p- & 2 o- & 2 m-Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 313.31): 313.10 (2.44), 222.00 (26.99), 153.00 (100.00), 125.05 (38.21), 91.05 (63.36), 69.00 (2.73); Elem. Anal. (%, for C16H15N3O4): Calculated (Found): C: 61.34 (61.39), H: 4.83 (4.80), N: 13.41 (13.41).● 5,5'-[5,5'-Methylenebis(1,3,4-oxadiazole-5,2-diyl)]di- benzene-1,2,3-triol (3s): Recryst. from DMF/abs. EtOH (2:1, v/v); Col. & App.: whitish buff crystalline powder; Yield: 84.0% (Conv.), 95.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3424 (O-H), 2922 (C-H, Arom.), 2850 (C-H, Aliph.), 1659 & 1651 (C=N), 1615 & 1548 & 1538 & 1504 & 1469 & 1455 (C=C, Arom.), 1285 & 1208 (C-O), Str. 1083 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 3.366-4.276 (s, 2H, CH2), 5.368 (s, 6H, 6 Arom. OH), 6.777-6.781 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 400.30): 400.10 (18.29), 109.10 (19.54), 83.10 (31.08), 69.10 (42.27), 57.05 (100.00), 55.00 (51.87); Elem. Anal. (%, for C17H12N4O8): Calculated (Found): C: 51.01 (51.09), H: 3.02 (3.03), N: 14.00 (14.04).● 5,5'-{5,5'-[(1R,2R)-1,2-Dihydroxyethane-1,2-diyl]bis(1,3, 4-oxadiazole-5,2-diyl)}dibenzene-1,2,3-triol (3t): Recryst. from DMF/abs. EtOH (2:1, v/v); Col. & App.: brownish orange fine powder; Yield: 75.0% (Conv.), 97.0% (MW); M.P.: 198-200 °C; IR (υ in cm-1): Str. & Bro. 3416 (O-H), Str. 2972 (C-H, Arom.), Str. 2850 (C-H, Aliph.), Str. 1682 & Str. 1616 (C=N), 1551 & 1538 & 1531 & 1515 & 1449 & 1398 (C=C, Arom.), Str. 1240 & 1186 (C-O), 1099 & Str. 1030 (N-N); 1H-NMR (CDCl3, δ in ppm): 3.655 (s, 2H, 2 Aliph. OH), 4.918-4.971 (d, J = 7 Hz, 2H, 2 CH), 5.375 (s, 6H, 6 Arom. OH), 7.259-7.272 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 446.32): 447.00 (9.91), 446.00 (7.73), 125.00 (27.95), 79.95 (100.00), 69.00 (42.61), 57.05 (73.13); Elem. Anal. (%, for C18H14N4O10): Calculated (Found): C: 48.44 (48.48), H: 3.16 (3.14), N: 12.55 (12.53).● 1,2,3-Tris[5-(3,4,5-trihydroxyphenyl)-1,3,4-oxadiazol-2- yl]propan-2-ol (3u): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brown crystalline powder; Yield: 90.0% (Conv.), 99.0% (MW); M.P.: 99-100 °C (dec.); IR (υ in cm-1): Str. & Bro. 3460 (O-H), 2917 (C-H, Arom.), 2850 (C-H, Aliph.), Str. 1637 (C=N), 1489 & 1389 (C=C, Arom.), 1262 (C-O), Str. 1071 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 2.495-2.507 (s, 4H, 2 CH2), 3.338 (s, 1H, Aliph. OH), 5.373 (s, 9H, 9 Arom. OH), 7.270 (s, 6H, 6 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 636.48): 636.00 (4.35), 443.00 (3.38), 125.00 (5.69), 69.00 (22.54), 63.95 (100.00), 57.05 (37.95); Elem. Anal. (%, for C27H20N6O13): Calculated (Found): C: 50.95 (50.96), H: 3.17 (3.16), N: 13.20 (13.21).● (E)-5-(5-Styryl-1,3,4-oxadiazol-2-yl)benzene-1,2,3-triol (3v): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brownish green crystalline powder; Yield: 73.0% (Conv.), 97.5% (MW); M.P.: 299-300 °C (dec.); IR (υ in cm-1): Str. & Bro. 3445 (O-H), Str. 3080 & Str. 3055 (=C-H, Alkene), Str. 2918 (C-H, Arom.), Str. 1641 (C=C, Alkene), 1578 (C=N), 1556 & Str. 1514 & Str. 1450 & Str. 1387 (C=C, Arom.), 1257 & 1230 & 1203 (C-O), Str. 1072 (N-N); 1H-NMR (DMSO-d6, δ in ppm): 5.351 (s, 3H, 3 Arom. OH), 6.787 (s, 2H, 2 Benzene-H), 6.955 & 7.029 (2 d, J = 15.1 Hz, 2H, trans HC=CH), 7.282-7.327 (m, 5H, 1 p- & 2 m- & 2 o-Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 296.28): 296.10 (0.83), 153.00 (100.00), 125.05 (38.21), 103.05 (6.93), 77.00 (7.06), 69.00 (2.73); Elem. Anal. (%, for C16H12N2O4): Calculated (Found): C: 64.86 (64.80), H: 4.08 (4.09), N: 9.46 (9.49).● (E)-5,5'-[5,5'-(Ethene-1,2-diyl)bis(1,3,4-oxadiazole-5, 2-diyl)]dibenzene-1,2,3-triol (3w): Recryst. from abs. MeOH; Col. & App.: light brown fine powder; Yield: 93.2% (Conv.), 98.8% (MW); M.P.: 288-290 °C (dec.); IR (υ in cm-1): Str. & Bro. 3425 (O-H), Str. 3077 & Str. 3054 (=C-H, Alkene), Str. 2926 (C-H, Arom.), Str. 1602 (C=C, Alkene), 1546 (C=N), 1505 & 1467 & 1413 & 1373 (C=C, Arom.), 1286 & 1206 (C-O), Str. 1078 & Str. 1057 (N-N); 1H-NMR (CDCl3, δ in ppm): 5.362 (s, 6H, 6 Arom. OH), 6.776 (s, 4H, 4 Benzene-H), 6.788 (d, J = 15.1 Hz, 2H, trans HC=CH); MS (m/z (Rel. Int. in %), M.Wt. = 412.31): 412.10 (19.36), 111.10 (22.38), 95.10 (20.60), 71.10 (53.82), 69.10 (42.27), 57.05 (100.00); Elem. Anal. (%, for C18H12N4O8): Calculated (Found): C: 52.43 (52.47), H: 2.93 (2.92), N: 13.59 (13.57).● 5-[5-(4-Bromophenyl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3x): Recryst. from abs. EtOH; Col. & App.: buff crystalline powder; Yield: 82.8% (Conv.), 96.0% (MW); M.P.: >300 °C; IR (υ in cm-1): Str. & Bro. 3476 (O-H), Str. 3066 (C-H, Arom.), 1637 (C=N), 1551 & 1517 & 1499 & 1374 (C=C, Arom.), 1189 (C-O), Str. 1079 (N-N), Str. 530 (C-Br); 1H-NMR (DMSO-d6, δ in ppm): 5.382 (s, 3H, 3 Arom. OH), 6.785-6.882 (s, 2H, 2 Benzene-H), 7.305-7.313 (m, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 349.14): 349.10 (8.90), 193.10 (10.12), 157.10 (10.74), 125.10 (17.33), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C14H9BrN2O4): Calculated (Found): C: 48.16 (48.12), H: 2.60 (2.63), N: 8.02 (8.06).● 5,5'-(1,3,4-Oxadiazole-2,5-diyl)dibenzene-1,2,3-triol (3y): Recryst. from abs. EtOH/H2O (3:1, v/v); Col. & App.: brown crystalline powder; Yield: 88.0% (Conv.), 98.2% (MW); M.P.: 82-84 °C; IR (υ in cm-1): Str. & Bro. 3466 (O-H), Str. 3090 (C-H, Arom.), 1647 (C=N), 1553 & 1533 & 1468 & 1380 (C=C, Arom.), 1199 (C-O), Str. 1071 (N-N); 1H-NMR (CDCl3, δ in ppm): 5.333 (s, 6H, 6 Arom. OH), 6.736 (s, 4H, 4 Benzene-H); MS (m/z (Rel. Int. in %), M.Wt. = 318.24): 318.10 (2.44), 193.00 (0.90), 153.00 (100.00), 125.05 (38.21), 69.00 (2.73), 57.00 (2.59); Elem. Anal. (%, for C14H10N2O7): Calculated (Found): C: 52.84 (52.91), H: 3.17 (3.15), N: 8.80 (8.76).● 5-[5-(7-Chloro-4-hydroxyquinolin-3-yl)-1,3,4-oxadiazol-2-yl]benzene- 1,2,3-triol (3z): Recryst. from hexane; Col. & App.: brownish buff fine powder; Yield: 86.4% (Conv.), 95.5% (MW); M.P.: 60-61 °C (dec.); IR (υ in cm-1): Str. & Bro. 3467 (O-H), Str. 3100 (C-H, Arom.), Str. 1620 (C=N), 1533 & 1483 & 1457 & 1378 (C=C, Arom.), Str. 1081 (C-O), Str. 983 (N-N), 869 (C-Cl); 1H-NMR (DMSO-d6, δ in ppm): 5.337 (s, 4H, 4 Arom. OH), 6.716 (s, 2H, 2 Benzene-H), 7.707 (dd, J = 7.5 Hz & 1.5 Hz, 1H, Quinoline-H-7), 7.949 (d, J = 1.5 Hz, 1H, Quinoline-H-9), 8.406 & 8.428 (d & s, JH-6 = 7.5 Hz, 2H, Quinoline-H-6,2); MS (m/z (Rel. Int. in %), M.Wt. = 371.73): 371.10 (9.51), 193.10 (10.12), 144.10 (9.20), 129.10 (28.07), 69.00 (52.45), 57.05 (100.00); Elem. Anal. (%, for C17H10ClN3O5): Calculated (Found): C: 54.93 (54.91), H: 2.71 (2.72), N: 11.30 (11.33).4.2. Biological Evaluation
4.2.1. ABTS Test
- All reagents and L-ascorbic acid were purchased from Aldrich Chemical Co., U.S.A.; while pure EtOH (of very high analytical grade) was purchased from El-Nasr Co. for Pharmaceutical Chemicals, Egypt. This assay was done according to the original idea of Re et al. [55] with very slight modifications.2,2'-Azinobis(3-ethylbenzothiazoline-6-sulfonic acid) radical cation (ABTS·+) is a free and stable radical cation which is able to react with any compound that can give a hydrogen atom or an electron (i.e., antioxidants, such as phenols and thiols), with decolorization of its dark green color. ABTS test is usually used to evaluate the antioxidant capacity of the biological fluids and many pure organic compounds.The ABTS·+ radical cation (blue-dark green) was prepared by reacting (i.e., mixing) equal volumes of ABTS stock solution (colorless; 7 mM in pure distilled H2O) and K2S2O8 stock solution (potassium persulfate; 3.5 mM in pure distilled H2O) (ABTS and K2S2O8 react stoichiometrically at a ratio of 2:1, respectively). The mixture was kept and allowed to stand in the dark at R.T. overnight (i.e., for about 12-16 h in the darkness) until the reaction was complete and the strong spectrophotometric absorbance (under UV) at a wavelength of 734 nm reaches the maximal stable value to obtain the ABTS·+ stock solution which is valid for use in this form for about 2-3 days when stored in the dark at R.T. The ABTS·+ working solution was prepared by diluting the ABTS·+ stock solution in pure EtOH to have an absorbance (Ablank) of 0.7±0.02 (after three times of measurement) at a wavelength of 734 nm and the solution was equilibrated with a temperature control set at 30 °C in an incubator (Ablank was adjusted in this present assay to be exactly 0.7 before measuring the absorbance for all the test compounds). Free radical scavenging activity was assessed by mixing 1.5 mL of the blue-green ABTS·+ working solution with 10 μL of the solutions of the target test compounds (3n-z) at various concentrations ranging from 10 to 300 µM (in distilled H2O, pure EtOH, or mixture of both of them according to the solubility of each compound). The change in absorbance at 734 nm was immediately monitored at 0, 0.5, 1 min after the addition (i.e., after the mixing) and again at 5 min intervals until a steady-state value was obtained. The steady state was achieved after 15 min in this present assay, so the absorbance value for each test compound after its addition to ABTS·+ solution (Atest) was taken after 15 min of their mixing. Values are means of three independent determinations (as all the measurements were taken three independent times after each period for each concentration of each test compound). The percent reduction in absorbance (which represents the ABTS·+ radical cation scavenging activity of the test compound) was calculated according to the following equation:ABTS·+ radical cation scavenging activity of test compound (%) = 100(Ablank-Atest)/Ablank.Where, Ablank or A0 is the absorbance of ABTS·+ radical cation in H2O and EtOH directly before reaction time = 0 min, i.e., directly before adding the test compound to the ABTS·+ radical cation (Ablank was adjusted to be 0.70), while Atest or A15 is the absorbance of the ABTS·+ radical cation (or of the reaction mixture) in H2O and EtOH at reaction time = 15 min, i.e., after 15 min of adding the test compound to the ABTS·+ radical cation. For each of the test compounds, the IC50 (the inhibitory concentration 50%, it is the concentration of any test compound needed to inhibit or reduce the absorption or the amount of ABTS·+ radical cations by 50% at a wavelength of 734 nm) was determined after 15 min of reaction and compared to that of L-ascorbic acid (taken as the reference and standard antioxidant compound in this assay). The antioxidant or anti-ABTS·+ IC50 value (for each test compound along with the reference L-ascorbic acid) was calculated using GraphPad Prism 6 software (U.S.A., 2015) and the lower the IC50 value is, the more powerful the test compound as antioxidant is (i.e., the stronger the antioxidant capacity of the test compound is).
4.2.2. DPPH Test
- DPPH, L-ascorbic acid, and trolox were purchased from Aldrich Chemical Co., U.S.A.; while pure EtOH (of very high analytical grade) was purchased from El-Nasr Co. for Pharmaceutical Chemicals, Egypt. This assay was done according to the procedure described by Prouillac et al. [56]. The ability of phenolic/thiol derivatives to donate a hydrogen atom was also evaluated by their ability to react with the stable 2,2-diphenyl-1-picrylhydrazyl free radical (DPPH·). DPPH· free radicals show a strong absorption band (dark red-violet color) at 516 nm and become colorless on reduction (i.e., their absorption band color fades away upon the absorption band reduction by a free radical scavenger compound). DPPH test is, exactly like ABTS test, used to evaluate the antioxidant capacity of the biological fluids and many pure compounds.The target test 1,3,4-oxadiazole derivatives (compounds 3n-z) were added at various concentrations, varying from 5 to 150 µM in pure EtOH or aqueous EtOH solution (according to the solubility of each test compound), to a freshly prepared ethanolic solution of DPPH· (80 μM). The absorbance was firstly measured at 516 nm at time = 0 min (i.e., directly before adding the solution of the test compound to the DPPH· solution) (A0) and after 30 min of incubation at R.T. (i.e., after 30 min of adding the test compound to the DPPH· solution and beginning the reaction) (A30). Values are means of three independent determinations (as all the experiments were carried out in triplicate) and the percent reduction in absorbance or the percent inhibition of DPPH· free radical (which represents the DPPH· free radical scavenging activity of the test compound) (I%) was calculated in the following way:I% = 100(A0-A30)/A0.For each of the test compounds, the IC50 (the inhibitory concentration 50%, it is the concentration of any test compound needed to inhibit or reduce the absorption or the amount of DPPH· free radicals by 50% at a wavelength of 516 nm) was determined after 30 min of reaction and compared to those of L-ascorbic acid and trolox (both were standardized as the reference antioxidant compounds in this assay). The antioxidant or anti-DPPH· IC50 value (for each of the test compounds along with the two references L-ascorbic acid and trolox) was calculated using GraphPad Prism 6 software (U.S.A., 2015) and the less the IC50 value is, the more potent and active the test compound as antioxidant is.
ACKNOWLEDGMENTS
- We are thankful to the staff members of the Departments of Pharmaceutical Organic Chemistry, Medicinal Chemistry, Pharmacognosy, and Pharmacology & Toxicology (Faculty of Pharmacy, Mansoura University, Mansoura, Egypt) for their support and interest in this new research. We are also very grateful to Mr. Ahmed A. Ibrahim (a laboratory technician at the Department of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt) for his technical help in the in vitro antioxidant assays.
Appendix
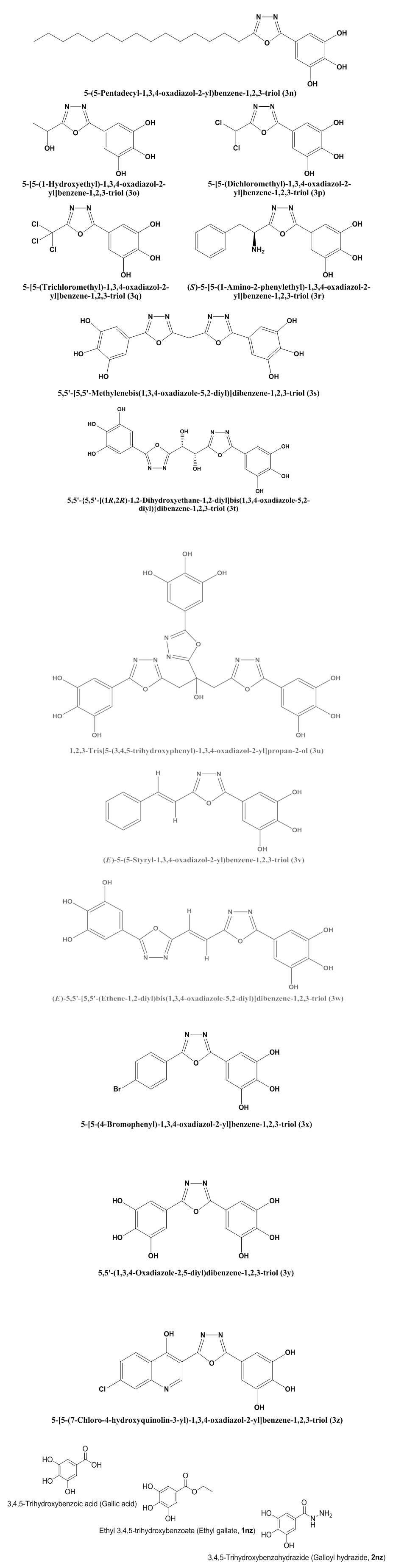 | Appendix A. Chemical Structures and IUPAC Nomenclature of All the Research Compounds |
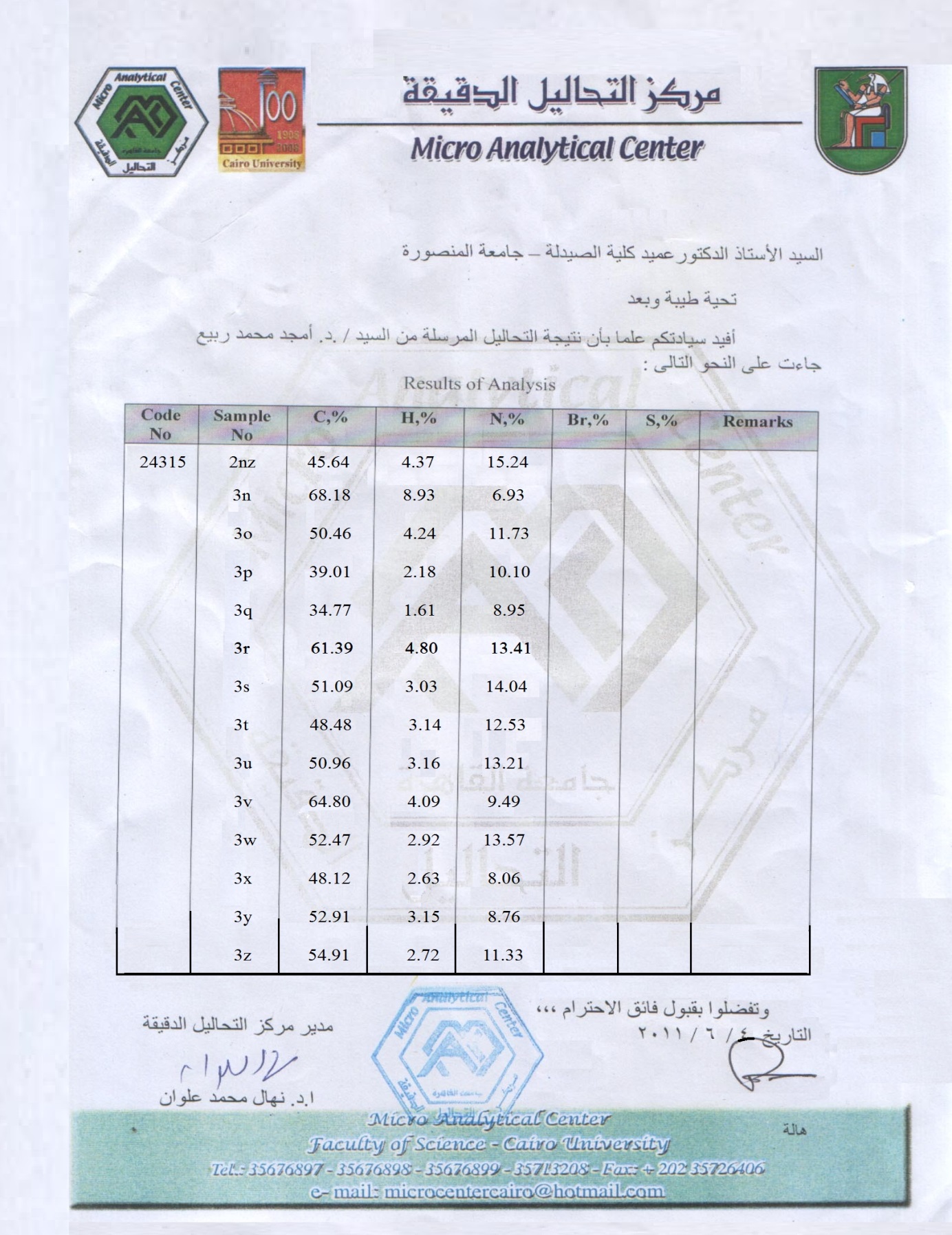 | Appendix B. Scanned Copy of the Elemental Analyses (Microanalyses of C, H, and N Contents) Results for Compound 2nz & Compounds 3n-z |
References
| [1] | Halliwell, B. Antioxidants: The basics-what they are and how to evaluate them. Adv. Pharmacol. 1996, 38(Part I), 3-20. |
| [2] | Halliwell, B.; Gutteridge, J. M. C. Free Radicals in Biology and Medicine; 4th Edition. Oxford University Press: New York, U.S.A., 2007. |
| [3] | Diplock, A. T.; Charleux, J.-L.; Crozier-Willi, G.; Kok, F. J.; Rice-Evans, C.; Roberfroid, M.; Stahl, W.; Viña-Ribes, J. Functional food science and defence against reactive oxidative species. Br. J. Nutr. 1998, 80(Suppl. 1), S77-S112. |
| [4] | Sies, H. Oxidative stress: Oxidants and antioxidants (in Physiological Society Symposium: Impaired endothelial and smooth muscle cell function in oxidative stress). Exp. Physiol. 1997, 82(2), 291-295. |
| [5] | Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58(1), 39-46. |
| [6] | Rahman, T.; Hosen, I.; Towhidul Islam, M. M.; Shekhar, H. U. Oxidative stress and human health. Adv. Biosci. Biotechnol. 2012, 3(7A), 997-1019, and references cited therein. |
| [7] | Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40(8), 959-975. |
| [8] | Sheu, S.-S.; Nauduri, D.; Anders, M. W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta - Mol. Basis Dis. 2006, 1762(2), 256-265. |
| [9] | Ng, F.; Berk, M.; Dean, O.; Bush, A. I. Oxidative stress in psychiatric disorders: Evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. 2008, 11(6), 851-876. |
| [10] | Pieczenik, S. R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83(1), 84-92. |
| [11] | Kuloglu, M.; Atmaca, M.; Tezcan, E.; Gecici, Ö.; Tunckol, H.; Ustundag, B. Antioxidant enzyme activities and malondialdehyde levels in patients with obsessive- compulsive disorder. Neuropsychobiology 2002, 46(1), 27-32. |
| [12] | Awasthi, D.; Church, D. F.; Torbati, D.; Carey, M. E.; Pryor, W. A. Oxidative stress following traumatic brain injury in rats. Surg. Neurol. 1997, 47(6), 575-582. |
| [13] | Alwis, D. S.; Johnstone, V.; Yan, E.; Rajan, R. Diffuse traumatic brain injury and the sensory brain. Proc. Aust. Physiol. Soc. 2013, 44, 13-26. |
| [14] | Kumar, S. V.; Saritha, G.; Fareedullah, M. Role of antioxidants and oxidative stress in cardiovascular diseases. Ann. Biol. Res. 2010, 1(3), 158-173. |
| [15] | Pinchuk, I.; Lichtenberg, D. The mechanism of action of antioxidants against lipoprotein peroxidation, evaluation based on kinetic experiments. Prog. Lipid Res. 2002, 41(4), 279-314. |
| [16] | Lakshmi, S. V. V.; Padmaja, G.; Kuppusamy, P.; Kutala, V. K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46(6), 421-440. |
| [17] | Shinde, A.; Ganu, J.; Naik, P.; Sawant, A. Oxidative stress and antioxidative status in patients with alcoholic liver disease. Biomed. Res. 2012, 23(1), 105-108. |
| [18] | Robles, L.; Vaziri, N. D.; Ichii, H. Role of oxidative stress in the pathogenesis of pancreatitis: Effect of antioxidant therapy. Pancreatic Disord. Ther. 2013, 3(1), 112. |
| [19] | Herrling, T.; Jung, K.; Fuchs, J. The role of melanin as protector against free radicals in skin and its role as free radical indicator in hair. Spectrochim. Acta, Part A 2008, 69(5), 1429-1435. |
| [20] | Franski, R. Biological Activities of the compounds bearing 1,3,4-oxa(thia)diazole ring. Asian J. Chem. 2005, 17(4), 2063-2075. |
| [21] | De Oliveira, C. S.; Lira, B. F.; Barbosa-Filho, J. M.; Lorenzo, J. G. F.; de Athayde-Filho, P. F. Synthetic approaches and pharmacological activity of 1,3,4-oxadiazoles: A review of the literature from 2000-2012. Molecules 2012, 17(9), 10192-10231. |
| [22] | Mehta, D. K.; Das, R. Synthesis and in vitro antioxidant activity of some new 2,5-disubstituted-1,3,4-oxadiazoles containing furan moiety. Int. J. Pharm. Sci. Res. 2011, 2(11), 2959-2963. |
| [23] | Yang, C. S.; Lambert, J. D.; Sang, S. Antioxidative and anticarcinogenic activities of tea polyphenols. Arch. Toxicol. 2009, 83(1), 11-21. |
| [24] | Pannala, A. S.; Chan, T. S.; O’Brien, P. J.; Rice-Evans, C. A. Flavonoid B-ring chemistry and antioxidant activity: Fast reaction kinetics. Biochem. Biophys. Res. Commun. 2001, 282(5), 1161-1168. |
| [25] | Rice-Evans, C. A.; Miller, N. J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biol. Med. 1996, 20(7), 933-956. |
| [26] | Takaoka, S.; Takaoka, N.; Minoshima, Y.; Huang, J.-M.; Kubo, M.; Harada, K.; Hioki, H.; Fukuyama, Y. Isolation, synthesis, and neurite outgrowth-promoting activity of illicinin A from the flowers of Illicium anisatum. Tetrahedron 2009, 65(40), 8354-8361, and references cited therein. |
| [27] | Majumdar, K. C.; Chattopadhyay, B.; Shyam, P. K.; Pal, N. A new disc-shaped mesogenic compound with olefinic linkage derived from triphenylamine: Synthesis, mesogenic behavior and fluorescence properties. Tetrahedron Lett. 2009, 50(49), 6901-6905, and references cited therein. |
| [28] | Ambika; Singh, P. P.; Chauhan, S. M. S. Chemoselective esterification of phenolic acids in the presence of sodium bicarbonate in ionic liquids. Synth. Commun. 2008, 38(6), 928-936. |
| [29] | Dodo, K.; Minato, T.; Noguchi-Yachide, T.; Suganuma, M.; Hashimoto, Y. Antiproliferative and apoptosis-inducing activities of alkyl gallate and gallamide derivatives related to (–)-epigallocatechin gallate. Bioorg. Med. Chem. 2008, 16(17), 7975-7982. |
| [30] | Łukasik, N; Wagner-Wysiecka, E. A review of amide bond formation in microwave organic synthesis. Curr. Org. Synth. 2014, 11(4), 592-604, and references cited therein. |
| [31] | Zheng, X.; Li, Z.; Wang, Y.; Chen, W.; Huang, Q.; Liu, C.; Song, G. Syntheses and insecticidal activities of novel 2,5-disubstituted 1,3,4-oxadiazoles. J. Fluorine Chem. 2003, 123(2), 163-169. |
| [32] | Chandrakantha, B.; Shetty, P.; Nambiyar, V.; Isloor, N.; Isloor, A. M. Synthesis, characterization and biological activity of some new 1,3,4-oxadiazoles bearing 2-fluoro-4-methoxyphenyl moiety. Eur. J. Med. Chem. 2010, 45(3), 1206-1210. |
| [33] | Mathew, V.; Keshavayya, J.; Vaidya, V. P.; Giles, D. Studies on synthesis and pharmacological activities of 3,6-disubstituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and their dihydro analogues. Eur. J. Med. Chem. 2007, 42(6), 823-840. |
| [34] | Amir, M.; Javed, S. A.; Kumar, H. Synthesis of some 1,3,4-oxadiazole derivatives as potential anti-inflammatory agents. Indian J. Chem. 2007, 46B(6), 1014-1019. |
| [35] | Saha, A.; Kumar, R.; Kumar, R.; Devakumar, C. Development and assessment of green synthesis of hydrazides. Indian J. Chem. 2010, 49B(4), 526-531, and references cited therein. |
| [36] | Demirbaş, N. Synthesis and characterization of new triheterocyclic compounds consisting of 1,2,4-triazol-3-one, 1,3,4-thiadiazole and 1,3,4-oxadiazole rings. Turk. J. Chem. 2005, 29(2), 125-133, and references cited therein. |
| [37] | Kempe, G.; Bögel, M.; Roewer, G. Constitution and redox stability of copper(II)-complexes with substituted hydrazines. J. Prakt. Chem. (Leipzig) 1981, 323(3), 360-366, and references cited therein. |
| [38] | Koufaki, M.; Kiziridi, C.; Alexi, X.; Alexis, M. N. Design and synthesis of novel neuroprotective 1,2-dithiolane/chroman hybrids. Bioorg. Med. Chem. 2009, 17(17), 6432-6441. |
| [39] | Arunkumar, S.; Ilango, K.; Manikandan, R. S.; Sudha, M.; Ramalakshmi, N. Synthesis, characterisation and biological evaluation of some novel 2,5-disubstituted-1,3,4-oxadiazole derivatives of gallic acid. Int. J. ChemTech Res. 2009, 1(4), 1094-1099. |
| [40] | Ilango, K.; Arunkumar, S. Synthesis, antimicrobial and antitubercular activities of some novel trihydroxybenzamidoazetidin-2-one derivatives. Trop. J. Pharm. Res. 2011, 10(2), 219-229. |
| [41] | Mandal, S. K.; Saha, D.; Jain, V. K.; Jain, B. Synthesis and antitubercular activity of some triazole derivatives of propyl gallate. Int. J. Pharma Sci. Res. 2010, 1(11), 465-473. |
| [42] | Mandal, S. K.; Saha, D.; Jain, B.; Jain, V. K. Synthesis, characterization and evaluation of antitubercular activity of some novel triazole derivatives of gallic acid. Int. J. Res. Pharm. Biomed. Sci. 2011, 2(1), 168-174. |
| [43] | Mandal, S. K.; Saha, D.; Jain, V. K.; Jain, B. Synthesis, characterization and evaluation of antibacterial and antifungal activity of triazole derivatives of gallic acid. Int. J. Appl. Biol. Pharm. Technol. 2010, I(3), 1300-1311. |
| [44] | Arunkumar, S.; Ilango, K.; Ravindar, B.; Ramalakshmi, N. Synthesis and biological evaluation of some novel triazolothiadiazole derivatives of gallic acid. Der Pharma Chem. 2009, 1(1), 70-77, and references cited therein. |
| [45] | Arunkumar, S.; Ilango, K.; Manikandan, R. S.; Ramalakshmi, N. Synthesis and anti-inflammatory activity of some novel pyrazole derivatives of gallic acid. E-J. Chem. 2009, 6(S1), S123-S128. |
| [46] | Ilango, K.; Arunkumar, S. Synthesis and antitubercular activity of novel thiazolidinone derivatives of gallic acid. J. Pharm. Res. 2011, 4(9), 3001-3003. |
| [47] | Padmavathi, V.; Reddy, G. S.; Padmaja, A.; Kondaiah, P.; Ali-Shazia. Synthesis, antimicrobial and cytotoxic activities of 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazoles. Eur. J. Med. Chem. 2009, 44(5), 2106-2112. |
| [48] | Gwaram, N. S.; Ali, H. M.; Abdulla, M. A.; Buckle, M. J. C.; Sukumaran, S. D.; Chung, L. Y.; Othman, R.; Alhadi, A. A.; Yehye, W. A.; Hadi, A. H. A.; Hassandarvish, P.; Khaledi, H.; Abdelwahab, S. I. Synthesis, characterization, X-ray crystallography, acetyl cholinesterase inhibition and antioxidant activities of some novel ketone derivatives of gallic hydrazide-derived Schiff bases. Molecules 2012, 17(3), 2408-2427. |
| [49] | Gohil, V. M.; Agrawal, S. K.; Saxena, A. K.; Garg, D.; Gopimohan, C.; Bhutani, K. K. Synthesis, biological evaluation and molecular docking of aryl hydrazines and hydrazides for anticancer activity. Indian J. Exp. Biol. 2010, 48(3), 265-268, and references cited therein. |
| [50] | Padmavathi, V.; Reddy, G. S.; Mohan, A. V. N.; Mahesh, K. Synthesis of symmetrical and unsymmetrical 1,3,4-oxadiazoles and their interconversion to 1,3,4-thiadiazoles and 1,2,4-triazoles. ARKIVOC (Gainesville, FL, U.S.) 2008, 2008(xvii), 48-60. |
| [51] | Mahajan, A.; Pai, N. Simultaneous isolation and identification of phytoconstituents from Terminalia chebula by preparative chromatography. J. Chem. Pharm. Res. 2010, 2(5), 97-103. |
| [52] | Silverstein, R. M.; Webster, F. X.; Kiemle, D. J. Spectrometric Identification of Organic Compounds; 7th Edition. John Wiley & Sons, Inc.: New York, U.S.A., 2005. |
| [53] | Kumar, H.; Javed, S. A.; Khan, S. A.; Amir, M. 1,3,4-Oxadiazole/thiadiazole and 1,2,4-triazole derivatives of biphenyl-4-yloxy acetic acid: Synthesis and preliminary evaluation of biological properties. Eur. J. Med. Chem. 2008, 43(12), 2688-2698. |
| [54] | Khan, M. T. H.; Choudhary, M. I.; Khan, K. M.; Rani, M.; Atta-ur-Rahman. Structure-activity relationships of tyrosinase inhibitory combinatorial library of 2,5-disubstituted-1,3,4-oxadiazole analogues. Bioorg. Med. Chem. 2005, 13(10), 3385-3395. |
| [55] | Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Biol. Med. 1999, 26(9-10), 1231-1237. |
| [56] | Prouillac, C.; Vicendo, P.; Garrigues, J.-C.; Poteau, R.; Rima, G. Evaluation of new thiadiazoles and benzothiazoles as potential radioprotectors: Free radical scavenging activity in vitro and theoretical studies (QSAR, DFT). Free Radical Biol. Med. 2009, 46(8), 1139-1148, and references cited therein. |