-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2016; 6(2): 39-53
doi:10.5923/j.ajoc.20160602.01

Synthesis, Characterization and in vitro Cytotoxic Evaluation of Some Novel Heterocyclic Compounds Bearing the Indole Ring
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAsmaa S. Salman1, Naema A. Mahmoud1, Mona A. Mohamed2, Anhar Abdel-Aziem1, Doaa M. Elsisi1
1Department of Chemistry, Faculty of Science, Al-Azhar University, Girls’ Branch, Nasr City, Cairo, Egypt
2Biochemistry Division, Faculty of Science, Al-Azhar University, Girls’ Branch, Nasr City, Cairo, Egypt
Correspondence to: Asmaa S. Salman, Department of Chemistry, Faculty of Science, Al-Azhar University, Girls’ Branch, Nasr City, Cairo, Egypt.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Reactions of 1H-indole-3-carboxaldhyde 1 with thiosemicarbazide derivatives give thiosemicarbazone derivatives 2a,b. Cyclization of thiosemicarbazone 2a with HCl, Ac2O, phenacyl bromides and chloroacetic acid afforded the corresponding 1,2,4-triazole-3-thiol 3, diacetyl derivative 4, 1,3-thiazole derivative 5 and 1,3-thiazolidin-4-one derivative 6, respectively. Compound 6 undergoes a series of heterocyclization reactions to give new heterocyclic compounds. Structures of the newly synthesized compounds have been confirmed by elemental analysis and spectral data. The newly synthesized compounds were evaluated for in vitro cytotoxic activity against three human cancer cell lines, including human liver cancer (Hep G2), human colon cancer (HT-29) and human breast cancer (MCF-7) using MTT assays.
Keywords: Thiosemicarbazone, 1.3-Thiazole, 1,3-Thiazoldinone, Pyrazolo [3,4-d]1,3-thiazole, Cytotoxic activity, MTT assay
Cite this paper: Asmaa S. Salman, Naema A. Mahmoud, Mona A. Mohamed, Anhar Abdel-Aziem, Doaa M. Elsisi, Synthesis, Characterization and in vitro Cytotoxic Evaluation of Some Novel Heterocyclic Compounds Bearing the Indole Ring, American Journal of Organic Chemistry, Vol. 6 No. 2, 2016, pp. 39-53. doi: 10.5923/j.ajoc.20160602.01.
Article Outline
1. Introduction
- Thiosemicarbazones has been used as intermediates for the preparation of many heterocyclic compounds. In the literature, manyresearchers have reported the S/N regioselective nucleophilic completion in the synthesis of heterocyclic compounds by intramolecular cyclization reactions. Changes in reaction conditions can induce S-attack or N-attack to eventually afford different cyclic products from a single starting material. Moreover, thiosemicarbazones bearing an aromatic heterocyclic moiety seem to possess enhanced biological activities [1, 2]. On other hand, heterocyclic compounds containing the indole ring are of major importance due to their therapeutic and pharmacological activities [3-7]. In view of the above and in continuation of our studies on the synthesis of heterocyclic compounds exhibiting biological activity, we report herein the successful reaction of 1H-indol-3-carboxaldehyde with thiosemicarbazide derivatives to afford the corresponding thiosemicarbazones derivatives. Subsequent cyclization by different reagents and different conditions gave novel heterocyclic compounds bearing the indole moiety which were then investigated for potential cytotoxic activities.
2. Experiment
2.1. General
- Melting points were measured on a Gallenkamp apparatus and are uncorrected. IR spectra were recorded on Shimadzu FT-IR 8101 PC infrared spectrophotometer (υmax in cm-1). The 1H-NMR and 13C NMR spectra were determined in DMSO-d6 at 300 MHz on a Varian Mercury VXR-300 NMR spectrometer using TMS as an internal standard. Mass spectra were measured on a GCMS-QP1000 EX spectrometer at 70 Ev. Elemental analyses were carried out at the Microanalyticalcenter of Cairo University and the main chemical warfare laboratories.
2.2. Chemistry
- 2.2.1. General Procedure for the Preparation of Thiosemicarbazones 2a,bAn equimolar mixture of2-(4-bromophenyl)-1H-indole-3-carboxaldehyde 1 and the selected thiosemicarbazide such as 4-(4-methylphenyl)-thiosemicarbazide or 4-(4- phenyl-1, 3-thiazol-2-yl) thiosemicarbazide (0.01 mol) were refluxed in absolute ethanol (20 mL) in the presence of 2-3 drops of glacial acetic acid for 3h. The reaction mixture was cooled to room temperature, the product separated and filtered, washed with cold water, dried and recrystallized from the appropriate solvent to give 2a,b.1-[2-(4-Bromophenyl)-1H-indol-3-ylmethylene]-N-(4-methylphenyl)thiosemicarbazone (2a)Yellow powder. Yield 80%, m.p. 249-250°C (ethanol-DMF). FT-IR (KBr, υmax/cm-1): 3135, 3317 (NH), 3042, 2975, 2857 (CH), 1246 (C=S). 1H-NMR (DMSO-d6) δ ppm: 2.32 (s, 3H, CH3), 7.17-7.28 (m, 4H, Ar-H), 7.45-7.52 (m, 4H, Ar-H), 7.60 (d, 1H, indole proton), 7.77-7.95 (m, 2H, indole proton), 8.33 (d, 1H, indole proton), 8.55 (s, 1H, =CH), 10.01 (s, 1H, NH exchanged by D2O), 11.46 (s, 1H, NH exchanged by D2O), 11.99 (s, 1H, NH exchanged by D2O). MS: m/z (%): 463 (M+, 0.3), 357 (1.3), 313 (0.8), 298 (72.5), 284 (10), 271 (100), 216 (23.3), 192 (16.3). Anal. calcd for C23H19BrN4S (463.39): C, 59.61; H, 4.13; Br, 17.24; N, 12.09; S, 6.92.Found: C, 59.41; H, 4.00; Br, 17.04; N, 12.00; S, 6.72.1-[2-(4-Bromophenyl)-1H-indol-3-ylmethylene]-N-(4-phenyl-1,3-thiazol-2-yl)-thiosemicarbazone (2b)Yellow powder. Yield 60%, m.p. 140-142°C (ethanol). FT-IR (KBr, υmax/cm-1): 3223, 3161, 3125 (NH), 3039, 2967, 2864 (CH), 1237 (C=S). 1H-NMR (DMSO-d6) δ ppm: 6.95-7.30 (m, 10H, Ar-H and H-5 thiazole), 7.32 (d, 1H, indole proton), 7.69-7.87 (m, 2H, indole proton), 8.19 (d, 1H, indole proton), 8.22 (s, 1H, N=CH), 8.47 (s, 1H, NH exchanged by D2O), 8.90 (s, 1H, NH exchanged by D2O), 12.44 (s, 1H, NH exchanged by D2O). Anal. calcd for C25H18 BrN5S2 (532.48): C, 56.39; H, 3.41; Br, 15.01; N, 13.15; S, 12.04. Found: C, 56.19; H, 3.21; Br, 14.89; N, 13.00; S, 11.89.2.2.2. 5-[2-(4-Bromophenyl)-1H-indol-3-yl]-4-(4- methyl phenyl)-4H-1,2,4–triazole-3-thiol (3)A solution of thiosemicarbazonederivative 2a (0.01 mol) in absolute ethanol (15 mL) containing a few drops of HCl was refluxed for 2h. After cooling and dilution with water, the solid formed were filtered off, washed with water, air dried and recrystallized from ethanol to give 3 as green powder. Yield 62%, m.p. 336–338oC (ethanol). FT-IR (KBr, υmax/cm-1): 3166 (NH), 3097, 2951, 2919 (CH), 1606 (C=N). 1H-NMR (DMSO-d6) δ ppm: 2.08 (s, 3H, CH3), 7.16-7.48 (m, 8H, Ar-H), 7.78 (d, 1H, indole proton), 7.65- 7.94 (m, 2H, indole proton), 8.43 (d, 1H, indole proton), 4.33 (s, 1H, SH exchanged by D2O), 12.05 (s, 1H, NH exchanged by D2O). 13C NMR (DMSO-d6) δ ppm: 20.44 (CH3), 154.84, 154.98 (2XC=N), 162.17 (C-S), 106.49, 111.47, 120.92, 121.00, 122.43, 122.70, 123.10, 125.81, 130.24, 131.14, 131.32, 131.93, 136.51, 141.73. Anal. calcd for C23H17BrN4S (461.38): C, 59.87; H, 3.71; Br, 17.32; N, 12.14; S, 6.95. Found: C, 59.57; H, 3.51; Br, 17.22; N, 12.04; S, 6.85.2.2.3. N-[4-Acetyl-5-(2-(4-bromophenyl)-1H-indol-3-yl)-4,5-dihydro-1,3,4-thiadiazol-2-yl]-N-(4-methylphenyl) acetamide(4)A solution of the thiosemicarbazone derivative 2a in acetic anhydride (12 mL) was heated under reflux for 5h with continuous stirring and then allowed to attain room temperature. The reaction mixture was slowly added to 400 mL of ice-cold water and then stirred at room temperature for 1h. The separated product was collected by filtration, washed with water, dried, and recrystallized from ethanol and DMF (2:1) to give 4 as orange powder, yield 55%, m.p. 180-182°C. FT-IR (KBr, υmax/cm-1): 3419 (NH), 3044, 2986, 2919 (CH), 1750, 1688 (2XC=O). 1H-NMR (DMSO-d6) δ ppm: 2.16 (s, 3H, CH3), 2.19 (s, 3H, CH3) , 2.26 (s, 3H, CH3), 6.93-7.28 (m, 9H, Ar-H and H-5, thiadiazole ring), 7.34 (d, 1H, indole proton), 7.51-7.82 (m, 2H, indole proton), 8.40 (d, 1H, indole proton), 11.55 (s, 1H, NH exchanged by D2O). MS: m/z, (%): 547 (M+, 0.5), 517 (0.3), 502 (0.2), 489 (1.2), 446 (0.23), 358 (79.7), 276 (1.6), 271 (100), 77 (80.3). Anal. calcd for C27H23BrN4O2S (547.47): C, 59.23; H, 4.23; Br, 14.60; N, 10.23; S, 5.86. Found: C, 59.03; H, 4.03; Br, 14.40; N, 10.03; S, 5.56.2.2.4. 2-(4-Bromophenyl)-3-[3-(4-methylphenyl)-4-phenyl-1,3-thiazol-2(3H)-ylidene]hydrazonomethyl-1H-indole (5)To a solution of thiosemicarbazone derivative 2a (0.01 mol) in absolute ethanol (20 mL) was added equimolar amounts of phenacyl bromide and anhydrous sodium acetate. The reaction mixture was heated under reflux for 6h with continuous stirring, then partially concentrated under reduced pressure and left to cool. The separated solid product was filtered off and recrystallized from ethanol to give 5 as a yellow powder. Yield 60%, m.p. 280-282°C. FT-IR (KBr, υmax/cm-1): 3122 (NH), 3028, 2947, 2826 (CH). 1H-NMR (DMSO-d6) δ ppm: 2.27 (s, 3H, CH3), 6.58 (s, 1H, H-5 thiazole ring), 7.14-7.28 (m, 13H, Ar-H), 7.44 (d, 1H, indole proton), 7.54-7.75 (m, 2H, indole proton), 8.30 (d, 1H, indole proton), 8.41 (s, 1H, N=CH), 11.85 (s, 1H, NH exchanged by D2O). MS: m/z, (%): 563 (0.33), 430 (0.3), 367 (0.32), 354 (0.36), 291 (0.96), 270 (0.56), 252 (60.32), 134 (35.86), 61(100). Anal. calcd for C31H23BrN4S (563.51): C, 66.07; H, 4.11; Br, 14.18; N, 9.94; S, 5.69. Found: C, 65.97; H, 4.00; Br, 14.00; N, 9.64; S, 5.49.2.2.5. 2-(4-Bromophenyl)-3-[3-(4-methylphenyl)–4-oxo-1,3-thiazolidin-2-ylidene]-hydrazonomethyl-1H-indole (6)A mixture of thiosemicarbazone derivative 2a (0.01 mol), chloroacetic acid (0.01 mol), and anhydrous sodium acetate (0.01 mol) in glacial acetic acid (20 mL) was heated under reflux for 8h with continuous stirring. The reaction mixture was left to cool and poured into ice-cold water, and the separated solid was filtered off, washed with water, dried, and recrystallized from DMF to give 6 as a yellow powder. Yield 70%, m.p. 340-342°C; FT-IR (KBr, υmax/ cm-1): 3273 (NH), 3044, 2959, 2861 (CH), 1703 (C=O), 1601 (C=N). 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 4.09 (s, 2 H, CH2), 7.19-7.31 (m, 8H, Ar-H), 7.45 (d, 1H, indole proton), 7.52-7.75 (m, 2H, indole proton), 7.90 (s, 1H, CH=N), 8.35(d, 1H, indole proton), 11.04 (s, 1H, NH exchanged by D2O). 13C-NMR (DMSO-d6) δ ppm: 20.65 (CH3), 32.16 (CH2), 152 (N=CH), 162 (N=C), 172 (C=O), 108.37, 111.78, 121.16, 122.42, 123.22, 125.76, 128.01, 129.40, 129.5, 129.61, 130.96, 131.13, 131.85, 132.51, 136.48, 138.06, 141.55. Anal. calcd for C25H19BrN4OS (503.41): C, 59.65; H, 3.80; Br, 15.87; N, 11.13; S, 6.37. Found: C, 59.35; H, 3.50; Br, 15.67; N, 11.00; S, 6.27.2.2.6. 2-(4-Bromophenyl)-3-[5-benzylidene-3-(4-methyl-phenyl)-4-oxo-1,3-thiazolidin-2-ylidene] hydrazonomethyl -1H-indole (7)To a solution of compound 6 (0.01 mol) and anhydrous sodium acetate (0.015 mol) in glacial acetic acid (10 mL) was added the benzaldehyde (0.01 mol). The mixture was heated under reflux for 6h with continuous stirring. The reaction mixture was left to cool and poured onto crushed ice with stirring. The separated solid was filtered off, washed with water, dried, and recrystallized from ethanol and DMF (2:1) to give 7 as orange powder. Yield 60%, m.p. 210-212°C. FT-IR (KBr) υmax/cm-1): 3292 (NH); 3029, 2942, 2842 (CH), 1683 (C=O). 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 7.25-7.32 (m, 14H, Ar-H and olefinic CH=), 7.51-7.77 (m, 3H, indole proton), 8.38 (d, 1H, indole proton), 8.42 (s, 1H, CH=N), 12.14 (s, 1H, NH exchanged by D2O). 13C-NMR (DMSO-d6) δ ppm: 20.68 (CH3), 142.00 (C=CH), 153.00 (N=CH), 156.00 (N=C), 165.00 (C=O), 108.12, 111.97, 122.27, 122.59, 125.72, 127.83, 128.09, 129.44, 129.60, 129.81, 131.06, 131.25, 131.80, 132.29, 133.84, 136.53, 138.38. Anal. calcd for C32H23BrN4OS (591.52): C, 64.98; H, 3.92; Br, 13.51; N, 9.47; S, 5.42. Found: C, 64.68; H, 3.72; Br, 13.31; N, 9.27; S, 5.22.2.2.7. 2-(4-Bromophenyl)-3-[6-(4-methyl phenyl)-2,3-diphenyl-2,3,3a,6-tetrahydro-5H-pyrazolo-[3,4-d]-1,3-thiazol-5-ylidene]-hydrazonomethyl-1H-indole (8)A mixture of compound 7 (0.01mol) and phenyl hydrazine (0.01 mol) was refluxed in ethanol (50 mL) in the presence of a few drops of acetic acid for 4h. The reaction mixture was cooled, and the solid separated was filtered off, washed with water and recrystallized from aqueous ethanol to give compound 8 as an orange powder. Yield 55%, m.p. 102-104°C. FT-IR (KBr, υmax/cm-1): 3229 (NH), 3054, 2936, 2857 (CH), 1605 (C=N). 1H-NMR (DMSO-d6) δ ppm: 2.24 (s, 3H, CH3), 4.09 (d, 1H, H-pyrazole), 6.67 (d, 1H, H-pyrazole), 7.08-7.22 (m, 18H, Ar-H), 7.24-7.94 (m, 3H, indole proton), 8.19 (d, 1H, indole proton), 8.52 (s, 1H, CH=N), 12.20 (s, 1H, NH exchanged by D2O). MS: m/z (%): 681 (M+, 0.1), 666 (0.4), 510 (3.2), 537 (2.2), 271 (100), 165 (73.5), 77 (30.9). Anal. calcd for C38H29BrN6S (681.65): C, 66.96; H, 4.29; Br, 11.72; N, 12.33; S, 4.70. Found: C, 66.76; H, 4.09; Br, 11.52; N, 12.03; S, 4.50.2.2.8. 2-(4-Bromophenyl)-3-[3-phenyl-6-(4-methylphenyl)-3,3a-dihydro-1,3-thiazolo[4,5-c]isoxazol-5-ylidene]-hydrazonomethyl-1H-indole (9)A mixture of compound 7 (0.01 mol), hydroxylamine hydrochloride (0.012 mol), sodium acetate (0.012 mol) was refluxed in ethanol (30 mL) in the presence of a few drops of acetic acid for 1h and kept overnight. Excess solvent was distilled off under reduced pressure and the remainder was then poured into water. The solid obtained was recrystallized from ethanol to give 9 as a white powder. Yield 65%; m.p. 170-172°C. FT-IR (KBr, υmax/cm-1): 3173 (NH); 3057, 2922, 2859 (CH). 1H-NMR (DMSO-d6) δ ppm: 2.17 (s, 3H, CH3), 4.35 (d,1H, H-isoxazole), 5.55 (d, 1H, H-isoxazole), 6.65-8.27 (m, 17 H, Ar-H and indole proton), 8.38 (s, 1H, CH= N), 11.57 (s, 1H, NH exchanged by D2O). MS: m/z (%): 606 (M+, 0.33), 530 (0.25), 488 (0.34), 324 (26.67), 297 (100), 271 (8.89). Anal. calcd for C32H24 BrN5OS (606.53): C, 63.37; H, 3.99; Br, 13.17; N, 11.55; S, 5.29. Found: C, 63.17; H, 3.79; Br, 13.00; N, 11.35; S, 5.09.2.2.9. 2-[2-(4-Bromophenyl)-1H-indol-3-ylmethylidene-hydrazono]-4-chloro-3-(4-methylphenyl)-2,3-dihydro-1,3-thiazole-5-carboxaldehyde (10)To the Vilsmeier-Haack complex prepared from DMF (10 mL) and POCl3 (0.02 mol) at 0°C was added the 1,3-thiazolidin-4-one derivative 6 (0.004 mol) and the reaction mixture was stirred at 60-65°C for 4h. The reaction mixture was kept overnight and it was then slowly added to crushed ice. The product separated on neutralization with NaHCO3, was filtered off and recrystallized from ethanol to give 10 as a yellow powder. Yield 70%; m.p. 150-152°C. FT-IR (KBr, υmax/cm-1): 3216 (NH), 3031, 2956, 2781 (CH), 1600 (C=N), 1675 (C=O). 1H-NMR (DMSO-d6) δ ppm: 2.08 (s, 3H, CH3), 7.19-8.22 (m, 12H, Ar-H and indole proton), 8.36 (s, 1H, CH=N), 9.95 (s, 1H, CHO), 12.45 (s, 1H, NH exchanged by D2O). 13C-NMR (DMSO-d6) δ ppm: 20.56 (CH3), 135.89 (C-Cl), 151 (N=CH), 156 (N=C), 164(C=O), 105.37, 121.01, 123.44, 125.71, 127.99, 129.40, 129.51, 131.20, 131.29, 131.32, 131.40, 131.88. Anal. calcd for C26H18BrClN4OS (549.87): C, 56.79; H, 3.30; Br, 14.53; Cl, 6.45; N, 10.19; S, 5.83. Found: C, 56.59; H, 3.00; Br, 14.33; Cl, 6.25; N, 10.00; S, 5.53.2.2.10. 2-(4-Bromophenyl)-3-[6-(4-methyl phenyl)-1,6-dihydro-5H-pyrazolo-[3,4-d]-1,3-thiazol-5-ylidene]- hydrazonomethyl-1H-indole (11)A mixture of compound 10 (0.01mol) and hydrazine hydrate (0.01 mol) was refluxed in ethanol (50 mL) for 4h. The reaction mixture was cooled, and the precipitate was filtered off and recrystallized from ethanol to give 11 as a yellow powder. Yield 64%, m.p. 300-302oC. FT-IR (KBr, υmax/cm-1): 3380, 3176 (NH), 3052, 2966, 2864 (CH), 1604 (C=N). 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 6.93-7.24 (m, 9H, Ar-H and H-3 pyrazole), 7.29 (d, 1H, indole proton), 7.54-7.85 (m, 2H, indole proton), 8.41 (d, 1H, indole proton), 8.90 (s, 1H, CH=N), 4.28 (s, 1H, NH exchanged by D2O), 12.03 (s, 1H, NH exchanged by D2O). MS: m/z (%): 527 (M+, 0.95), 567 (0.99), 281 (3.33), 254 (1.11), 248 (35.86), 118 (100). Anal. calcd for C26H19 BrN6S (527.44): C, 59.21; H, 3.63; Br, 15.15; N, 15.93; S, 6.08. Found: C, 59.00; H, 3.43; Br, 15.00; N, 15.63; S, 6.00.2.2.11. N'-{2-[2-(4-Bromophenyl)-1H-indol-3-ylmethyl-enehydrazono]-4-chloro-3-(4-methylphenyl)-2,3-dihydro-1,3-thiazol-5-ylmethylene}-2-cyanoacetohydrazide (12)An equimolar mixture of 10 (0.02 mol) and cyanoacetic acid hydrazide (0.02 mol) in absolute ethanol (30 mL) was heated under reflux for 2h. The precipitate formed after cooling was filtered off, washed with cold ethanol, dried and recrystallized from xylene to give 12 as an orange powder. Yield 50%; m.p. 230-232°C. FT-IR (KBr, υmax/ cm-1): 3327 (NH), 2920, 2853 (CH), 2196 (CN), 1668 (C=O). 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 4.22 (s, 2H, CH2), 7.15-7.27 (m, 8H, Ar-H), 7.38-7.92 (m, 3H, indole proton), 8.18 (d, 1H, indole proton), 8.29 (s, 1H, CH=N), 8.36 (s, 1H, CH=N), 11.33(s, 1H, NH exchanged by D2O), 11.49 (s, 1H, NH exchanged by D2O). MS: m/z (%): 630 (M+, 0.87), 538 (1.19), 383 (8.04), 348 (1.32), 270 (88.70), 295(100). Anal.calcd for C29H21BrClN7OS (630.95): C, 55.20; H, 3.35; Br, 12.66; Cl, 5.62; N, 15.54; S, 5.08. Found: C, 55.00; H, 3.15; Br, 12.46; Cl, 5.52; N, 15.34; S, 5.00.2.2.12. 2-[2-(4-Bromophenyl)-1H-indol-3-ylmethylene hydrazono]-3-(4-methylphenyl)-3,4–dihydro-1,3-thiazolo-[4,5-b]-1,5-benzodiazepine (13)An equimolar mixture of compound 10 (0.02 mol), o-phenylenediamine (0.02 mol) and 0.2 mL TEA in absolute ethanol (30 mL) was heated under reflux for 8h. The precipitate formed after cooling was filtered off, washed with cold ethanol, dried, and recrystallized from ethanol to give 13 as an orange powder. Yield 67%; m.p. 250-252°C. FT-IR (KBr, υmax/ cm-1): 3337 (NH), 3055, 2923, 2865 (CH). 1H-NMR (DMSO- d6) δ ppm: 2.27 (s, 3H, CH3), 7.22-8.37 (m, 17 H, Ar-H and benzodiazepine), 8.90 (s, 1H, CH= N), 12.03 (s, 1H, NH exchanged by D2O), 12.31(s, 1H, NH exchanged by D2O). MS: m/z (%): 603 (0.98), 504 (0.32), 334 (3.89), 316 (1.21), 308 (74.55), 281 (16.62), 245 (3.05), 77 (100). Anal. Calcd for C32H23BrN6S (603.53): C, 63.68; H, 3.84; Br, 13.24; N, 13.92; S, 5.31. Found: C, 63.48; H, 3.54; Br, 13.14; N, 13.62; S, 5.11.2.2.13. 2'-[2-(4-Bromophenyl)-1H-indol-3-ylmethylene hydrazono]-3'-(4-methylphenyl)-3-phenyl-2,5'–bis-1,3- thiazolidin-2'-ylidene-4,4'-dione 16To a stirred solution of 0.56g KOH (0.01mol) in 20 mL DMF, 1,3-thiazolidin-4-one 6 (0.10 mol) was added. After stirring for 30 min, phenyl isothiocyanate (0.01mol) was added to the resulting mixture and the reaction mixture stirred at room temperature for 12h. Then, ethyl chloroacetate (0.01 mol) was added to the reaction mixture and stirred for 6h. The reaction mixture was poured into crushed ice. The resulting precipitate was filtrated off, dried, and recrystallized from xylene to give 16 asanorange powder. Yield, 60%, m.p. 290-292°C. FT-IR (KBr, υmax/cm-1): 3267 (NH), 3042, 2964, 2919 (CH), 1702 (C=O), 1600 (C=N). 1H-NMR (DMSO-d6) δ ppm: 2.36 (s, 3H, CH3), 4.09 (s, 2H, CH2), 7.07-7.32 (m, 13H, Ar-H), 7.45-7.55 (m, 2H, indole proton), 7.72 (d, 1H, indole proton), 8.33 (s, 1H, CH= N), 8.35 (d, 1H, indole proton), 12.02 (s, 1H, NH exchanged by D2O). 13C-NMR (DMSO-d6) δ ppm: 20.75 (CH3), 32.16 (CH2), 152.65 (CH=N), 157.15 (C=N), 162.38 (C=O), 164.72 (C=O), 99.43, 108.37, 110.45, 111.82, 114.23, 122.42, 125.70, 126.87, 129.66, 129.94, 130.66, 130.74, 130.98, 131.96, 136.49, 138.06, 141.55, 149.95. Anal. Calcd for C34H24BrN5O2S2 (678.62): C, 60.18; H, 3.56; Br, 11.77; N, 10.32; S, 9.45. Found: C, 60.00; H, 3.36; Br, 11.57; N, 10.22; S, 9.25.2.2.14. 2-Oxo-2-phenylethyl{2-[2-(4-bromophenyl)-1H-indol-3-ylmethylenehydrazono]-3-(4-methylphenyl)-4-oxo}-1,3-thiazolidine-5-carbodithioate (18)To a stirred suspension of finely powdered potassium hydroxide (0.02 mol) in dry DMF (20 mL), 1,3- thiazolidin-4-one 6 (0.01 mol) was added. The resulted mixture was cooled at 10oC in an ice bath; then (0.01mol) carbon disulfide was added slowly over the course of 10 min. After complete addition, stirring of the reaction mixture was continued for 6h. Then, phenacyl bromide (0.01mol) was added to the mixture and stirring continued for 3h, then the mixture was poured into crushed ice and HCl. The resulting precipitate was filtrated off, dried, and recrystallized from xylene to give 18 as a red powder. Yield, 60%; m.p. 200-202°C. FT-IR (KBr, υmax/cm-1): 3274 (NH), 3056, 2967, 2861 (CH), 1702 (C=O), 1241 (C=S). 1H-NMR (DMSO- d6) δ ppm: 2.37 (s, 3H, CH3), 4.09 (s, 2H, CH2), 4.76 (s, 1H , H-5thiazolidinone), 7.13-7.75 (m, 13H, Ar-H), 7.39-7.97 (m, 3H, indole proton), 8.22 (d, 1H, indole proton), 8.38 (s, 1H, CH=N), 12.09 (s, 1H, NH exchanged by D2O). 13C-NMR (DMSO-d6) δ ppm: 10.36 (CH3), 30.01 (CH2), 147.39 (CH=N), 150.43 (C=N), 164.36 (C=O), 164.73 (C=O), 185.40 (C=S), 107.37, 110.00, 111.22, 114.03, 122.12, 124.60, 128.51, 130.52, 130.60, 130.65, 130.84, 130.85, 132.92, 133.08, 149.01. Anal. Calcd for C34H25 BrN4O2S2 (665.62): C, 61.35; H, 3.79; Br, 12.00; N, 8.42; S, 9.63. Found: C, 61.15; H, 3.59; Br, 11.89; N, 8.22; S, 9.43.
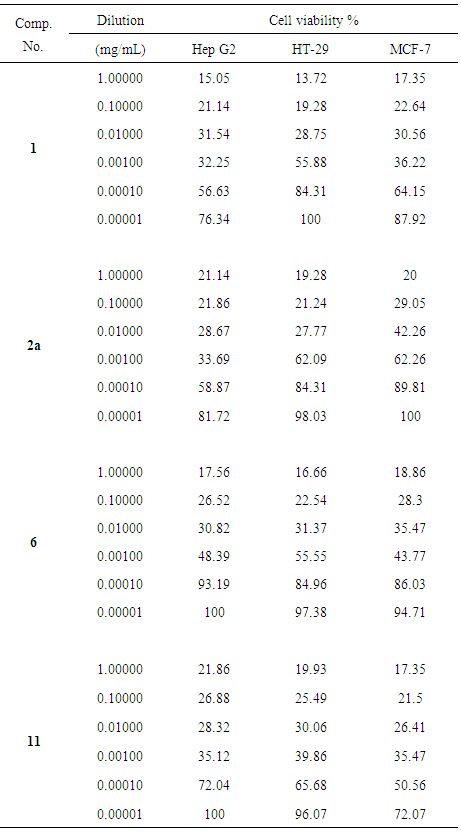
2.3. In Vitro Cytotoxic Screening (MTT assay)
- In vitro cytotoxicity of newly synthesized compounds 1, 2a, 6 and 11 were evaluated against human liver cancer cell (Hep G2), human colon cancer cell (HT-29) and human breast cancer cell (MCF-7) cell line using a standard MTT assay. The monolayer cells were detached with trypsin- ethylenediaminetetra-acetic acid (EDTA) to make singlet cell suspensions and viable cells were counted using a hemocytometer, then diluted with the fetal bovine serum (FBS) medium with 5% FBS to give final density of 2×105 cells/mL.). One hundred microliters per well of cell suspension were seeded into 96-well plates at a plating density of 10,000 cells/well and incubated to allow for cell attachment at 37°C, 5% CO2, 95% air and 100% relative humidity. The synthesized samples were dissolved in 1 mL dimethylsulfoxide (DMSO) and further diluted in serum free medium to produce six concentrations starting from 1 to 10-6 mg/mL. About 500-10,000 cells in 200 µl media per well were incubated at 37°C and 5% CO2 overnight to allow the cells to attach to the wells. 100 µl from each dilution of tested samples was added to each well, mixed by shaking at 150 rpm for 5 minutes, incubated at 37°C and 5% CO2 for 48h. 20 μl of MTT (5 mg/mL) in phosphate buffered saline (PBS) was added to each well plate and mixed by shaking at 150 rpm for 5 min and incubated at 37°C and 5% CO2 for 5h to allow the MTT to be metabolized. The medium with MTT was then flicked off and the formed formazan crystals (MTT metabolic product) were solubilized in 200 μl of DMSO and then absorbance was measured at 560 nm using a micro plate reader [8]. The viability of treated cells was calculated in reference to the untreated control cells by using the following formula:Cell viability (%) = [100 × (Sample Abs)/(Control Abs)].
3. Results and Discussion
3.1. Chemistry
- The synthetic procedures adopted to obtain the target compounds are outlined in Schemes 1-3. The key intermediate 1-[1H-indol-3-ylmethylene]thiosemicarbazone derivatives 2a,b were prepared by the reaction of 1H-indole-3-carboxaldehyde 1 with thiosemicarbazide derivatives such as 4-(4-methylphenyl)thiosemicarbazide or 4-(4-phenyl- 1,3-thiazol-2-yl)thiosemicarbazide in refluxing ethanol containing acetic acid [9] (Scheme 1). The structures of compounds 2a and 2b were based on analytical and spectral data. The 1H-NMR spectra of 2a displayed three D2O- exchangeable NH proton signals at δ 10.01, δ 11.46 and δ 11.99 ppm and a singlet signal at δ 2.32 ppm for the CH3 protons.
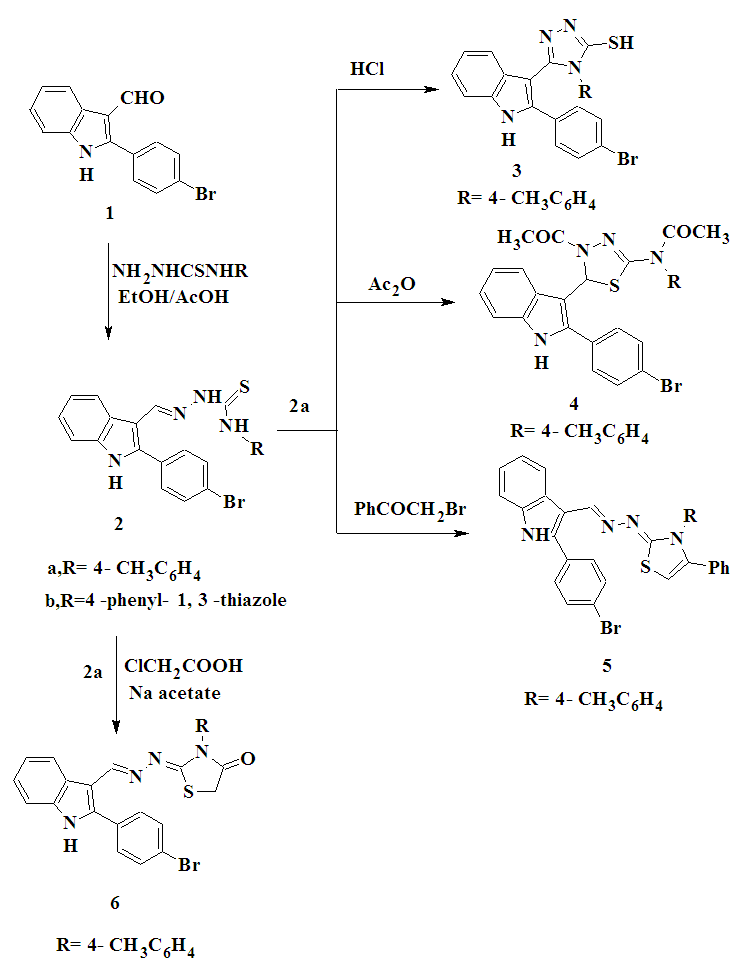 | Scheme 1. Synthesis of compounds 2-6 |
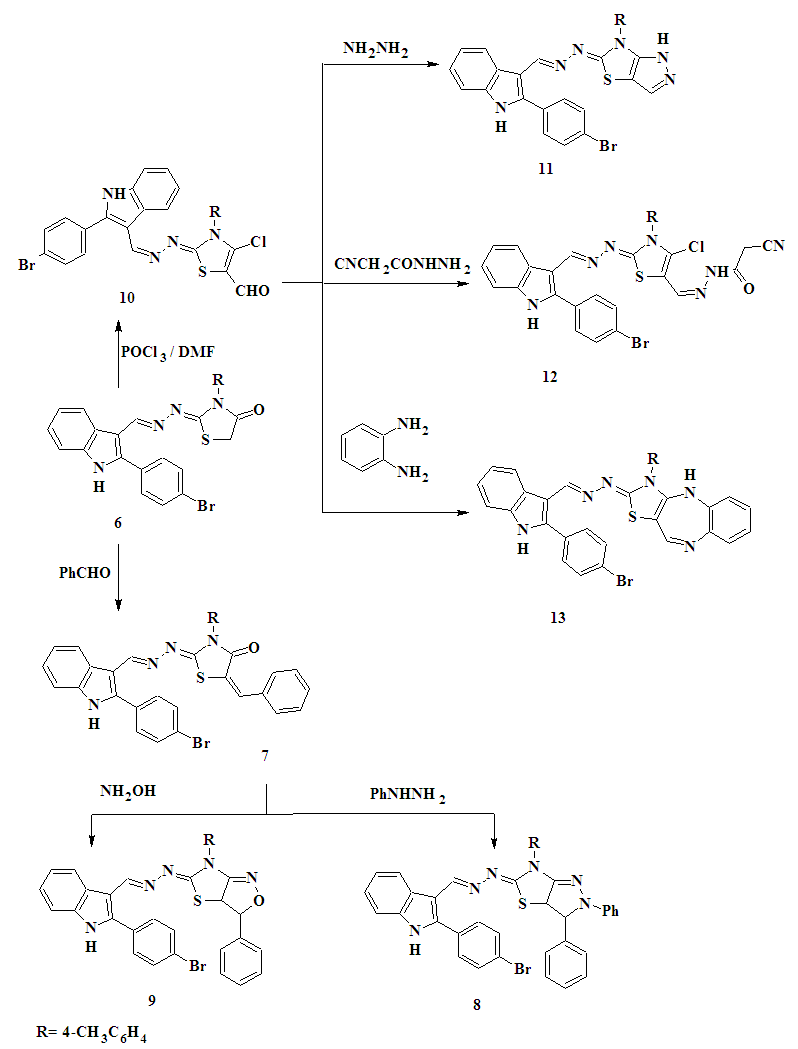 | Scheme 2. Synthesis of compounds 7-13 |
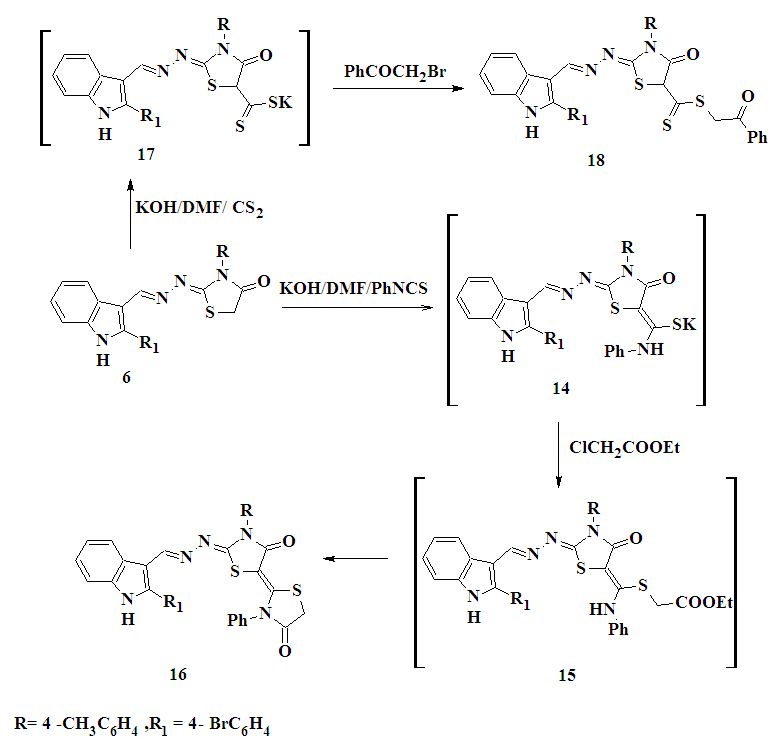 | Scheme 3. Synthesis of compounds 16 and 18 |
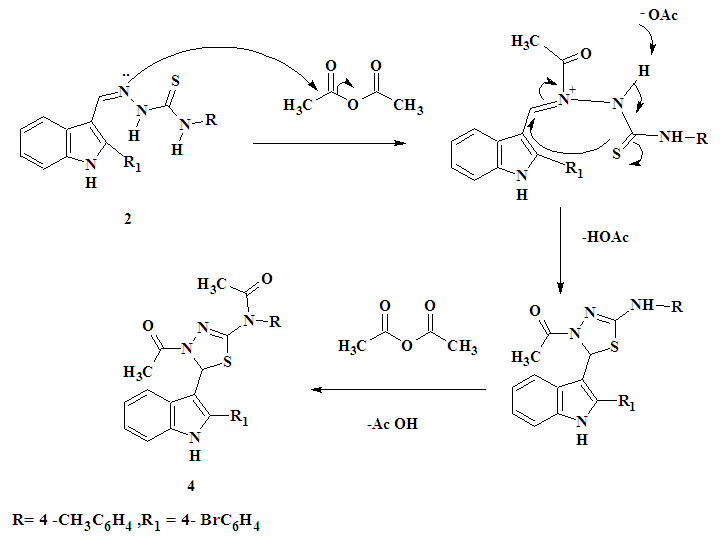 | Figure 1. Proposed mechanism for the formation of compound 4 |
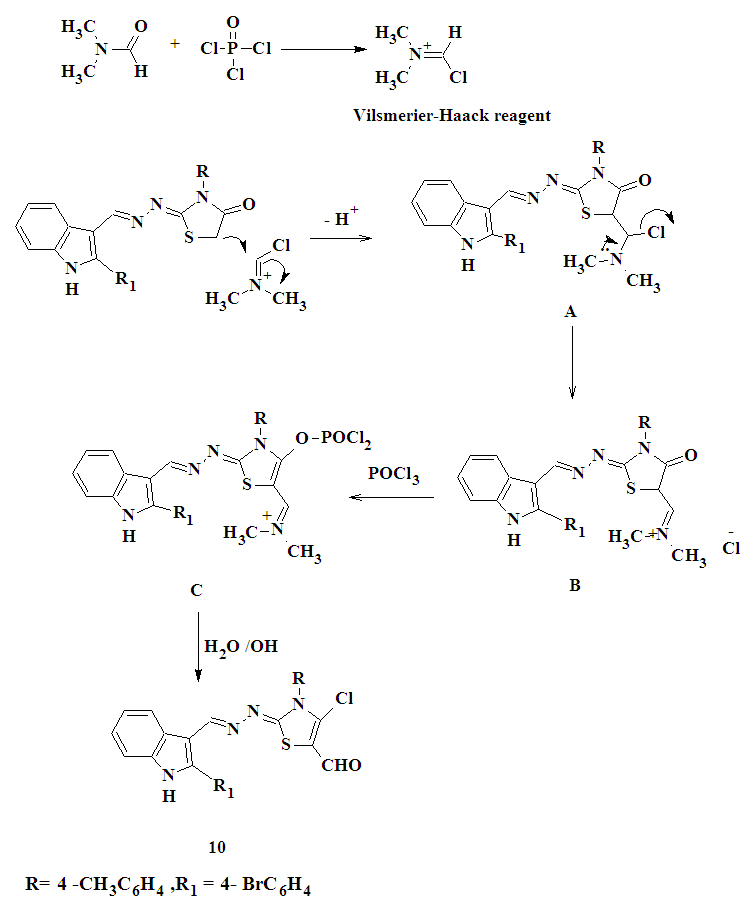 | Figure 2. Proposed mechanism for the formation of compound 10 |
3.2. In Vitro Cytotoxicity Screening
- The newly synthesized compounds 1, 2a, 6 and 11 were evaluated for their in vitro cytotoxic effects against human liver cancer (Hep G2) cell line, human colon cancer (HT-29) cell line and human breast cancer (MCF-7) cell line by the standard MTT (3- (4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay [22, 23]. The method is based on the ability of a mitochondrial dehydrogenase from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form purple formazan crystals which are impermeable to cell membranes (Scheme 4). The crystals can be solubilized by detergents. The number of living cells is directly proportional to the level of formed formazan which can be quantified photometrically. When the amount of purple formazan produced by cells treated with an agent is compared with the amount of formazan produced by untreated control cells, the effectiveness of the agent in causing death of cells can be deduced (see Figure 3).
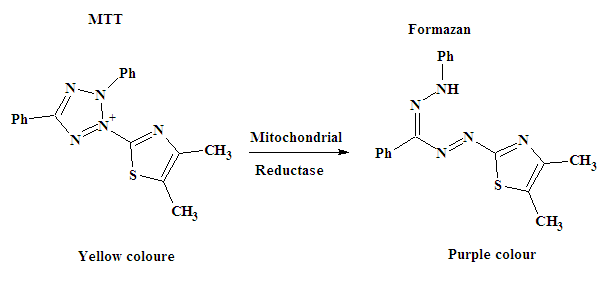 | Scheme 4. Principle of MTT assay |
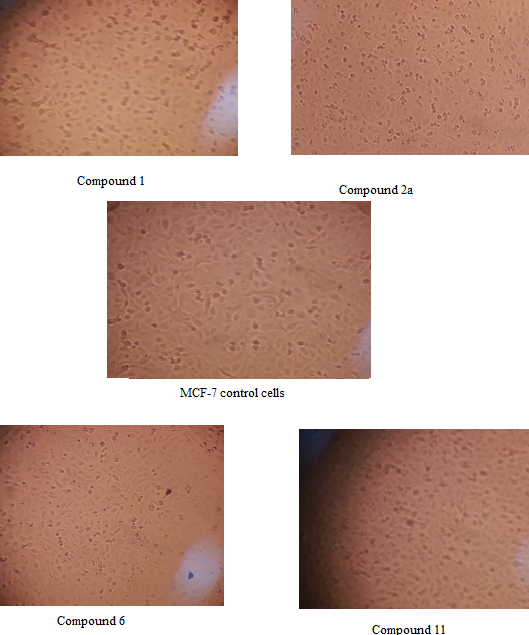 | Figure 3. Pictorial view change in MCF-7 cell morphology after exposure to MTT |
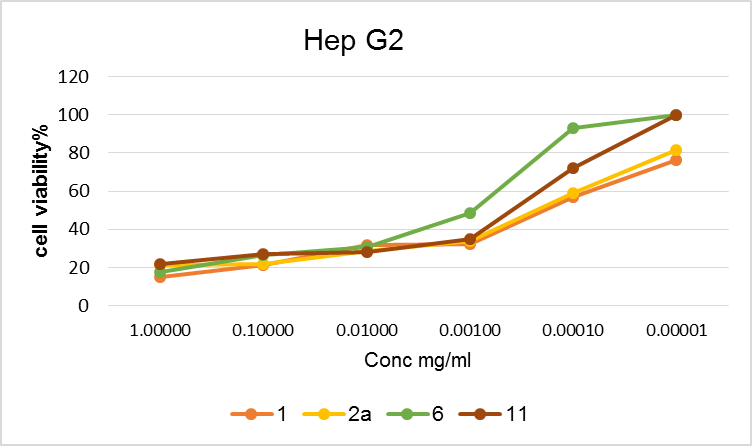 | Figure 4. Cell viability % of Hep G2 with different concentrations of the tested compounds |
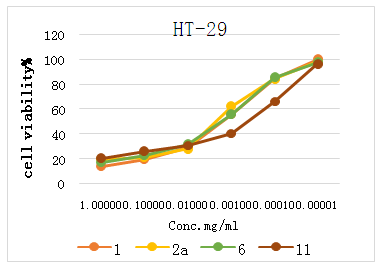 | Figure 5. Cell viability % of HT-29 with different concentrations of the tested compounds |
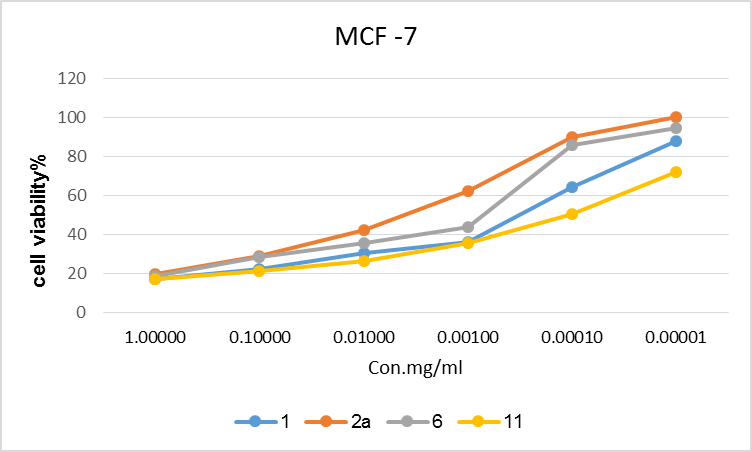 | Figure 6. Cell viability % of MCF-7 with different concentrations of the tested compounds |
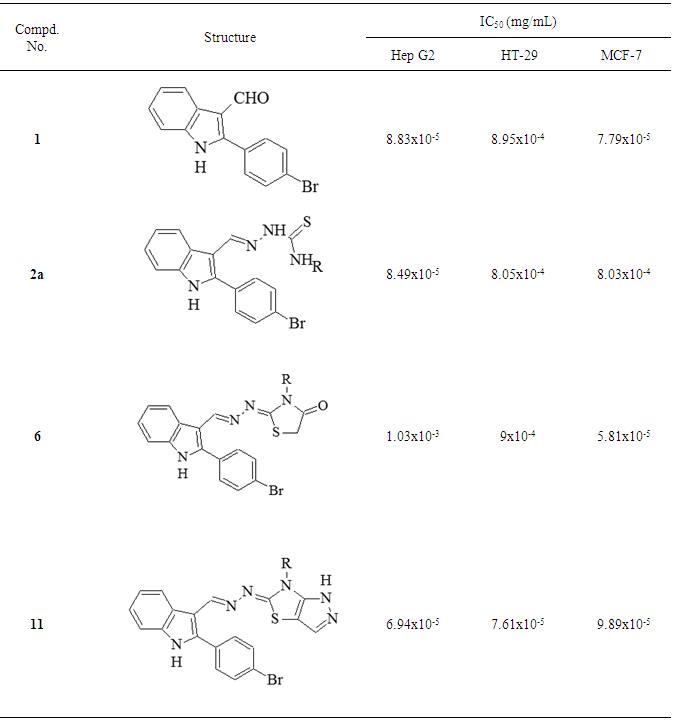
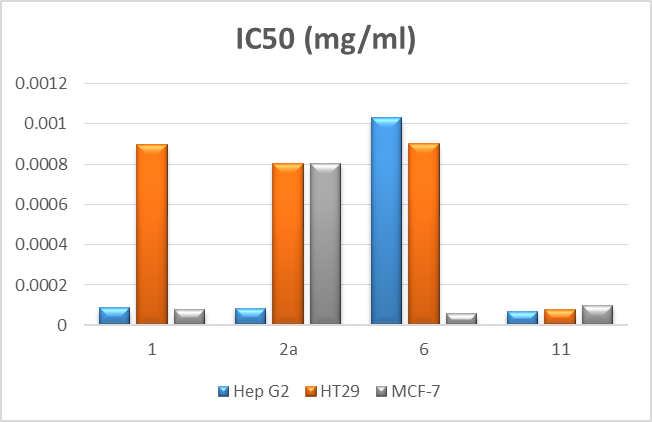 | Figure 7. Evaluation of IC50 of test compounds |
|
|
4. Conclusions
- In this work, a variety of heterocyclic systems have been synthesized from thiosemicarbazone derivatives. The newly synthesized compounds 1, 2a, 6 and 11 have been evaluated for in vitro cytotoxic activity against human liver cancer (Hep G2), human colon cancer (HT-29) and human breast cancer (MCF-7) cell lines using an MTT assay protocol. Compound 11 showed the best cytotoxic activity against all the three cancer cell lines due to the presence of pyrazolo [3,4-d]-1,3-thiazol group at position-3 of the in dole ring. Compound 1 also showed higher cytotoxic activities against the human liver cancer (Hep G2) and human breast cancer (MCF-7) cell line due to the presence of a CHO group at position-3 of the indole ring. Compound 2a also showed higher cytotoxic activities against the human liver cancer (Hep G2) cell line due to the presence of a thio- semicarbazone group at position-3 of the indole ring. Compound 6 also showed higher cytotoxic activities against the human breast cancer (MCF-7) cell line due to the presence of a 1,3-thiazolidine ring at position-3 of the indole ring. Hence, it can be suggested that compound 1, 2a, 6 and 11 could be used as leads in the design and development of new anticancer drugs.