-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2016; 6(1): 29-38
doi:10.5923/j.ajoc.20160601.04

Synthesis, Modeling and Anticonvulsant Activity of Some Phthalazinone Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRezkRezk A. Ayyad1, 2, Helmy Sakr1, Kamal El-Gamal2, 3
1Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Al-Azhar University, Nasr City, 11884, Cairo, Egypt
2Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Delta University, Gamasa, Dakahlia, Egypt
3Faculty of Pharmacy, Department of Organic Chemistry, Al-Azhar University, Nasr City, 11884, Cairo, Egypt
Correspondence to: RezkRezk A. Ayyad, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, Al-Azhar University, Nasr City, 11884, Cairo, Egypt.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A series containing 4-(4-chlorophenyl)-(2H)-phthalazinone moiety has been synthesized in good yield and evaluated for their anticonvulsant activities. The anticonvulsant evaluation was carried out using pentylenetetrazole (PTZ) induced-convulsions mice model and phenobarbitonesodium as a standard. Computer aided drug design (CADD) studies were performed to rationalize the best fitting value of the prepared compounds. The result is compatible with the reported SAR studies the best fitting value was noticed for compound 9b, which revealed the highest anticonvulsant activity.
Keywords: 4-(4-chlorophenyl)-(2H)-phthalazinone, AMPA-R, Anticonvulsant activity
Cite this paper: RezkRezk A. Ayyad, Helmy Sakr, Kamal El-Gamal, Synthesis, Modeling and Anticonvulsant Activity of Some Phthalazinone Derivatives, American Journal of Organic Chemistry, Vol. 6 No. 1, 2016, pp. 29-38. doi: 10.5923/j.ajoc.20160601.04.
Article Outline
1. Introduction
- Epilepsy is a syndrome, not a disease characterized byparoxysmal, excessive and hypersynchronous discharges of large number of neurons and it’s one of the most common neurologic diseases, is characterized by seizures, evoked by unexpected high level cranial neuronal discharges [1]. Approximately1% of the world’s population (~50 million people) is affected by epilepsy used anti-convulsants [2] that cause some adverse reactions such as drowsiness, ataxia, gastrointestinal disturbance, hepatotoxicity and megaloblastic anaemia [3-4], as well as life-threatening conditions in some rare cases [5]. Therefore, there is a crucial need for safer and more effective antiepileptic therapy.Nitrogen-containing heterocycles are indispensable structural units for medicinal chemists. Among the various heterocyclic compounds, Phthalazine form an attractive biologically active molecule as anticonvulsant agents [6–10]. Also they are known to possess other biological activities such as antidiabetic [11], antihypertensive [12] analgesic and anti-inflammatory [13], antiviral [14], antihistaminic [15], and potent antitumor [16] activities. Currently available clinically effective antiepileptic act by inducing prolonged inactivation of the Na+ channel, blocking Ca2+channel currents, or enhancing inhibitory GABAergic neurotransmission or via antagonism of glutamatergic neurotransmission [17]. In fact, the clinical use of NMDA-R antagonists is limited due to the unacceptable central side effects such as hallucination and ataxia [18] also trigger schizophrenia-like symptoms, perceptual alterations, and memory impairment [19]. The antagonists of (AMPA) receptor possess some advantages compared to (NMDA) receptor antagonists, including higher neuroprotective potency after ischemic attacks, higher anticonvulsant potency in temporal lobe epilepsy and reduced side effects (psychotomimetic action and impairment of learning and memory) [20-22]., most of AMPA-R antagonists characterized by high affinity and selectivity were build based on Phthalazine scaffold. Several series of 1,2- dihydrophthalazine derivatives were prepared andproved to possess anticonvulsant activity via antagonism of AMPA receptors in non-competitive manner. Representative examples of anticonvulsant Phthalazine compounds are shown in Figure 1; (1), (2) and (3) have been reported to exhibit anticonvulsant activity in animal models [23–25].
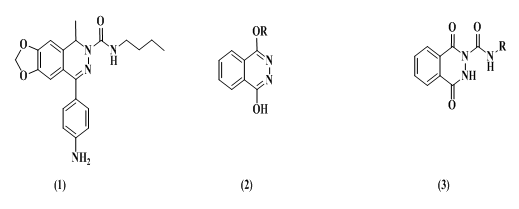 | Figure 1 |
2. Experimental
2.1. Chemistry
- Melting points were measured in capillary tube on a Graffin melting point apparatus and are uncorrected. The IR spectra were recorded on PyeUnicam SP 1000 IR spectrophotometer using KBr discs (λmax in cm-1).1H NMR spectra were performed either on a Jeol ECA (500 MHz) or Gemini 300BB (300MHz) and 300 MHz for 13C NMR) spectrometer, using TMS as internal standard and DMSO-d6 as solvent; the chemical shifts are reported in ppm (δ) and coupling constant (J) values are given in Hertz (Hz). Signal multiplicities are represented by s (singlet), d (doublet), t (triplet), q (quadruplet), and m (multiplet). All of the new compounds were analyzed for C, H and N and agreed with the proposed structures within ±0.4% of the theoretical values by the automated CHN analyser at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. Mass spectra were recorded on Hewlett Packard 5988 spectrometer at the RCMB. The purity of the compounds was checked by thin layer chromatography (TLC) on Merck silica gel 60 F254 precoated sheets. 4-(4-chlorophenyl)-(2H)-phthalazinone (2) were prepared adopting the reported procedures [27-30].2.1.1. 4-(4-Chlorophenyl)-2-(EthoxyCarbonylpropyl)-(2H)-Phthalazinone (3)A mixture of 4-(4-chlorophenyl)-(2H)-phthalazinone (2)[27-30](6.66 gm, 0.03 mole ) and anhydrous K2CO3 (8.28 gm, 0.06 mole) in dry acetone (30 ml) was refluxed for about one hour, then ethyl 4-bromobutyrate (4.3 ml, 0.03 mole) and KI (0.5 gm) were added to the reaction mixture which allowed to reflux and stirring for 16 hours. The acetone was evaporated and the residue was dissolved in water, neutralized with acetic acid and extracted with chloroform (3 x 30 ml). The chloroformic layer was washed with 10% aqueous sodium hydroxide and brine, dried over anhydrous sodium sulfate and then evaporated to dryness. The obtained solid product was crystallized from ethyl alcohol 95%.Yield: 83%; mp 80-81°C;IR (KBr, ν, cm-1):3047 (C-H aromatic), 1712 (C=O ester), 1644 (C=O phthalazine ring), 1570 (C=N).1H NMR (300 MHz, [D6]CDCl3): δ = 1.20 (t, 3H, CH3CH2O-), 2.2 (m, 2H, -CH2CH2CH2-), 2.45 (t, 2H, -CH2CH2COO-), 4.08 (t, 2H, -NCH2CH2-), 4.3 (q, 2H, - OCH2CH3), 7.2-8.5 (m, 8H, aromatic protons). 13C NMR (75 MHz, [D6] DMSO): δ = 14.4, 23.9, 31.1, 40.9, 61.2, 123.7, 126.6, 127.2, 128.4, 128.4, 130.3, 130.5, 130.5, 131.1, 132.2, 132.3, 134.8, 136.8, 158.2, 173.1. MS (m/z): 372 (M+2, 16.58%), 370 (M+, 16.17%), 327 (M-OC2H5, 7.27%), 299(M-COOC2H5, 3.23%), 269 (M– CH2CH2COOC2H5, 16.36%), 255 (M–C3H6COOC2H5 41.67%), and base beak at 242 (M–NC3H6COOC2H5, 100%). Anal.Calcd. For C20H19ClN2O3: C, 64.78; H, 5.16; N, 7.55. Found: C, 65.12; H, 5.28; N, 7.83.2.1.2. 2-(HydrazinoCarbonylPropyl)-4-(4-Chlorophenyl)-(2H)-Phthalazinone(4)Compound 4 was prepared via refluxing 2-(Ethoxycarbonylpropyl)-4-(4-chlorophenyl)-(2H)-phthalazinone (3) (6.5gm, 0.02 mole) in an excess of hydrazine hydrate 98% (15ml, 0.05 mol) for 4 hours. The reaction mixture was cooled to room temperature then poured on 250 g of crushed ice where pale yellow precipitate was produced. The precipitate was collected by filtration, dried over anhydrous potassium carbonate, washed with ether (3 x 20 mL), dried and crystallized from water as colourless crystals.Yield: 89%; mp149-150°C; IR (KBr, ν, cm-1): 3298 (NH2), 1650 (C=O hydrazide), 1539 (C=N). 1H NMR (300MHz, CDCl3): δ = 2.2 (m, 2H, -CH2CH2CH2-), 2.4 (t, 2H, -CH2CH2CO-), 3.9 (t, 2H, -NCH2CH2-), 4.3 (s, 2H, NH2 exchangeable with D2O), 7.2-7.8 (m, 9H, aromatic protons), 8.5 (s, H, NH exchangeable with D2O). 13C NMR (75 MHz, [D6] DMSO): δ = 22.2, 35.1, 41.3, 123.7, 126.2, 127.5, 129, 129, 130.3, 130.5, 130.5, 131.1, 132.2, 134.8, 136.8, 158.2, 176.5. MS (m/z): 356 (M+, 26.29%), 358 (M+2, 14.33%) and base beak at 325. Anal.Calcd. For C18H17ClN4O2: C, 60.59; H, 4.80; N, 15.70. Found: C, 61.02; H, 5.16; N, 15.92.2.1.3. 2-(substitutedBenzylidenehydrazinocarbonylpropyl)-4-(4-Chlorophenyl)-(2H)-Phthalazinone (5a-h)General procedureTo a stirred solution of 2-(Hydrazinocarbonylpropyl)-4-(4-chlorophenyl)-((2H)-phthalazinone (4) (1gm, 0.031 mole) in hot absolute ethyl alcohol (10 ml), the appropriate aromatic aldehyde (0.031 mol) and few drops of glacial acetic acid were added. The reaction mixture was refluxed for 3 hours. This reaction product is formed and precipitated during the reflux process. The formed precipitate was collected via filtration while hot, washed with ethyl alcohol (3 x 5 ml), dried and crystallized from suitable solvent (chloroform or absolute ethanol).2.1.4. 2-(benzylidene Hydrazine CarbonylPropyl)-4-(4-Chlorophenyl)-(2H)-Phthalazinone (5a)Yield: 83%; mp160-162°C; IR (KBr, ν, cm-1): 3187 (NH), 3036 (C-H aromatic), 2962(C-H aliphatic), 1655 (C=O) 1572 (C=N).1HNMR (300MHz, CDCl3): δ = 2.2 (m, 2H, -CH2CH2CH2-), 2.9 (t, 2H, -CH2CH2CO-), 4.4 (t, 2H, -NCH2CH2-), 7.2-7.8 (m, 13H, aromatic protons), 8.09 (s, 1H, N=CH) and 11.1 (s, 1H, NH exchangeable with D2O). 13C NMR (75 MHz, [D6] DMSO): δ = 22.2, 35.7, 41.2, 123.6, 126.2, 127.5, 129, 129, 129.2, 129.2, 129.5, 129.5, 130.3, 130.5, 130.5, 131.1, 131.1, 132.2, 133.8, 134.8, 136.5, 144.1, 158.2, 166.5. MS (m/z): 444 (M+, 0.09%), 443 (M-1, 0.14%) and base beak at 103(M –C3H6CONHN=CHC6H5, 100%). Anal.Calcd. For C25H21ClN4O2: C, 67.49; H, 4.76; N, 12.59. Found: C, 67.58; H, 4.64; N, 12.74.2.1.5. 2-{N-(4-Florobenzylidene Hydrazino Carbonyl Propyl)}-4-(4-Chlorophenyl)- (2H)-Phthalazinone (5b)Yield: 84%; mp185-186°C; IR (KBr, ν, cm-1): 3188 (NH), 3028 (C-H aromatic), 2962 (C-H aliphatic), 1658 (C=O) and 1591 (C=N).1HNMR (300MHz, CDCl3): δ = 2.27 (m, 2H, -CH2CH2CH2-), 2.71 (t, 2H, -CH2CH2CO-), 4.27 (t, 2H, -NCH2CH2-), 7.11-7.9 ( m, 12H, aromatic protons ), 8.06 (s, 1H, N=CH) and 11.99 (s, 1H, NH). MS (m/z): 463 (M+, 0.09%), 462 (M - 1, 0.14 %) and base beak at 129.9 (M–C3H6CONHN=CHC6H4F, 100%). Anal.Calcd. For C25H20ClFN4O2: C, 64.87; H, 4.35; N, 12.10. Found: C, 65.20; H, 4.48; N, 12.23.2.1.6. 2-{N-(4-N,N-Dimethylamino-)Benzylidenehydrazinocarbony lpropyl)}-4-(4-Chlorophenyl)-(2H)-Phthalazinone (5c)Yield: 82%; mp165-167°C; IR (KBr, ν, cm-1):3180 (NH), 3039 (C-H aromatic), 2962 (C-Haliphatic), 1716 (C=O), 1561 (C=N). 1HNMR, (300MHz, CDCl3) δ = 2.27 (m, 2H, -CH2CH2CH2-), 2.71 (t, 2H, -CH2CH2CO-), 4.27 (t, 2H, -NCH2CH2-), 7.11-7.9 (m, 12H, aromatic protons), 8.06 (s, 1H, N=CH ) and 11.99 (s, 1H, NH).13C NMR (75 MHz, [D6] DMSO): δ= 22.2, 35.5, 41.4, 41.4, 41.4, 111.9, 111.9, 123.3, 123.8,126.3, 127.8, 128.2, 128.2, 128.9, 128.9, 130.4, 130.7, 130.7, 131, 131.2, 132.2, 134.5, 136.8, 144.2, 153.5, 158.2, 167. MS (m/z): 487 (M+, 0.14 %), 489 (M + 2, 0.91%) and base beak at 76.10 (M –C12H10C1N3HNN=CHC6H4NC2H6, 100%). Anal.Calcd. For C27H26ClN5O2: C, 66.46; H, 5.37; N, 14.35. Found: C, 66.62; H, 5.49; N, 14.50.2.1.7. 2-{N-(4-Nitrobenzylidene Hydrazino carbonylpropyl)}-4-(4-Chlorophenyl) - (2H)-Phthalazinone (5d)Yield: 87%; mp190-192°C; IR (KBr, ν, cm-1): 3148(NH), 3092 (C-H aromatic), 296 (C-H aliphatic), 1672 (C=O), 1591 (C=N).). 1HNMR (300 MHz, CDCl3): δ = 2.1 (m, 2H, -CH2CH2CH2-), 2.76 (t, 2H, -CH2CH2CO-), 4.25 (t, 2H, -NCH2CH2-), 7.4-7.9 (m, 12H, aromatic protons), 8.10 (s, 1H, N=CH) and 11.2 (s, 1H, NH). Anal.Calcd. For C25H20ClN5O4: C, 61.29; H, 4.11; N, 14.30. Found: C, 61.43; H, 4.30; N, 14.48.2.1.8. 2-{N-(4-Methoxybenzylidene Hydrazino carbonylpropyl)}-4-(4-Chlorophenyl) (2H)-Phthalazinone (5e)Yield: 81%; mp160-163°C;IR (KBr, ν, cm-1):3290 (NH), 3035(C-Haromatic), 2930 (C-Haliphatic), 1715 (C=O), 1617 cm-1 (C=N).13C NMR (75 MHz, [D6] DMSO): δ= 22.2, 35.7, 41.3, 56, 114.5, 114.5, 123.8, 126, 126.3, 127.6, 129, 129, 130.3, 130.3, 130.5, 130.8, 130.8, 131.1, 131.2, 131.4, 134.8, 136.8, 144.2, 158.3, 162.9, 166.9. MS (m/z): 474 (M+, 0.23 %), 476 (M+2, 0.40 %) and base beak at 75.85. Anal.Calcd. For C26H23ClN4O3: C, 65.75; H, 4.88; N, 11.80. Found: C, 65.88; H, 5.10; N, 11.88.2.1.9. 2-{N-(3, 4, 5-Trimethoxybenzylidene Hydrazino carbonyl propyl)}-4-(4-Chlorophenyl) (2H)-Phthalazinone (5f)Yield: 84%; mp230-232 °C; IR (KBr, ν, cm-1):3180 (NH), 3002 (C-H aromatic), 2930 (C-H aliphatic), 1657 (C=O), 1574 (C=N). 1HNMR (300MHz, CDCl3): δ = 2.3 (m, 2H, -CH2CH2CH2-), 2.9 (t, 2H, -CH2CH2CO-), 3.88 (s, 9H, 3-OCH3), 4.44 (t, 2H, -NCH2CH2-), 7.27-7.86 (m, 10H, aromatic protons), 8.04 (s, 1H, N=CH) and 9.97 (s, 1H, NH). Anal.Calcd. For C28H27ClN4O5: C, 62.86; H, 5.09; N, 10.47. Found: C, 63.12; H, 5.18; N, 10.56.2.1.10. 2-{N-(4-Chloro)Benzylidenehydrazino carbonyl propyl)}-4-(4-Chlorophenyl) (2H)-Phthalazinone (5g)Yield: 81%; mp170-172°C; IR (KBr, ν, cm-1):3192 (NH), 3078 (C-H aromatic), 2953(C-Haliphatic), 1663 (C=O), 1592 cm-1 (C=N). 13C NMR (75 MHz, CDCl3): δ= 22.2, 35.5, 41.4, 123.8, 126.3, 126.9, 127.3, 127.5, 129, 129,130.1, 130.5, 130.6, 130.6, 131, 131.2, 132.3, 132.4, 134, 134.6, 134.6, 138.7, 158.3, 167.1. MS (m/z): 480 (M+2, 0.17%), 478 (M+, 0.38%), 479 (M - 1, 0.23%) and base beak at 88.65. Anal.Calcd. For C25H20Cl2N4O2: C, 62.64; H, 4.21; N, 11.69. Found: C, 62.79; H, 4.34; N, 11.87.2.1.11. 2-{N-(2,6-Dichlorobenzylidene Hydrazino carbonylpropyl)}-4-(4-Chlorophenyl) (2H)-Phthalazinone (5h)Yield: 77%; mp180-182°C;IR (KBr, ν, cm-1):3190 (NH), 3072 (C-Haromatic), 2947(C-Haliphatic), 1662 (C=O) and 1593 (C=N). MS (m/z): 512 (M+, 0.32%), 514 (M +2, 0.33%), 516 (M + 4, 0.16%) and base beak at 162.90. Anal.Calcd. For C25H19Cl3N4O2: C, 58.44; H, 3.73; N, 10.90. Found: C, 58.71; H, 3.89; N, 11.12.2.1.12. 4-(4-Chlorophenyl)-2-{[3-(5-Sulphanyl-[1, 3, 4]-Oxadiazol-2-yl)Propyl] Phthalazin-1(2H)-One (6)A mixture of compound 4 (3.56 g, 0.01 mol), carbon disulphide (0.76 gm, 0.71ml, 0.01 mole) and potassium hydroxide (0.56 gm, 0.01 mole) were refluxed in ethanol (20 ml) for 16 hours. The reaction mixture was cooled, filtered, boiled till clear solution and acidified with 20 ml 1N HCl. The solid obtained was filtered, washed with water several times and crystallized from absolute ethanol. Yield: 83%; mp210-211°C; IR (KBr, ν, cm-1): 3080 (C-H aromatic), 2940 (C-H aliphatic), 2774 (SH), 1619 (C=O amidic). 1HNMR (300MHz, [D6] CDCL3): δ = 2.39 (m, 2H, -CH2CH2CH2-), 2.84 (t, 2H, -CH2CH2CO-), 4.46 (t, 2H, -NCH2CH2-), 7.26 -7.85 (m, 8H, aromatic protons) and 11.41 (s, 1H, SH exchangeable with D2O). MS (m/z): 398 (M+, 0.54%), 400 (M+2, 0.24%) and base beak at 162 (100%). Anal.Calcd. For C19H15ClN4O2S: C, 57.21; H, 3.79; N, 14.05. Found: C, 57.40; H, 3.94; N, 14.33.2.1.13. Potassium salt of-5-[3-(4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl) Propyl)]-1, 3, 4-Oxadiazole-2-Thiolate(7)A clear solution of 4-(4-Chlorophenyl)-2-{[3-(5- sulphanyl-[1,3,4]-oxadiazol-2-yl)propyl]phthalazin-1(2H)-one (6) (3.98 gm, 0.01 mole) and potassium hydroxide (0.56 gm, 0.01 mole) was refluxed in absolute ethanol (20 ml) for 2 hours. Upon cooling to the r. t., a white precipitated product was obtained which was collected and washed with diethyl ether and air dried. Yield: 90%; mp> 300°C.2.1.14. 4-(4-Chlorophenyl)-2-{[3-(5-Methylsulphanyl-[1, 3, 4]-Oxadiazol-2-yl) Propyl]}-(2H)-Phthalazinone (8a)Equimolar quantity of the potassium salt (7) (4.36 gm, 0.01 mole) and methyl iodide (0.6 ml, 0.01 mole) in ethylalcohol (20 ml) was heated on water bath for 3 hrs. The reaction mixture was poured onto ice water (200ml) and stirred for 30 minutes. The obtained solid was filtered and crystallized from absolute ethanol.Yield: 80%; mp120-121°C; IR (KBr, ν, cm-1): 3031 (C-H aromatic), 2946 (C-H aliphatic), 1647 (C=O amidic). 1HNMR (300MHz, CDCl3): δ = 2.4 (m, 2H, -CH2CH2CH2-), 2.64 (s, 3H, S-CH3), 2.97 (t, 2H, -CH2CH2CO-), 4.37 (t, 2H, -NCH2CH2-), and 7.27-7.82 (m, 8H, aromatic protons). MS (m/z): 398 (M+, 0.54 %), 400 (M+2, 0.24%) and base beak at 162 (100%). Anal.Calcd. For C20H17ClN4O2S: C, 60.22; H, 4.80; N, 14.05. Found: C, 60.63; H, 4.95 N, 14.19.2.1.15. 4-(4-Chlorophenyl)-2-{[3-(5-Ethylsulphanyl-[1, 3, 4]-Oxadiazol-2-yl) propyl]}-(2H)-Phthalazinone(8b)Equimolarquantity of the potassium salt (7) (4.36 gm, 0.01 mole) and ethyl iodide (0.8 ml, 0.01 mole) in ethyl alcohol (20 ml) were heated on water bath for 3 hours. The reaction mixture was poured onto ice water (200 ml) and stirred for 30 min. The obtained solid was filtered and crystallized from absolute ethanol. Yield: 80%; mp89-90°C; IR (KBr, ν, cm-1):3057 (C-H aromatic), 2948 (C-H aliphatic), 1645 (C=O amidic). MS (m/z): 427 (M+, 16.48%), 429 (M+2, 3.68%) and base beak at 162 (100%). Anal.Calcd. For C21H19ClN4O2S: C, 59.08; H, 4.49; N, 13.12. Found: C, 59.42; H, 4.88 N, 13.60.2.1.16. 4-(4-Chlorophenyl)-2-{[3-(5-Allylsulphanyl-[1, 3, 4]-Oxadiazol-2-yl)Propyl]}-(2H)-Phthalazinone (8c)Equimolar quantity of the potassium salt (7) (4.36 gm, 0.01 mole) and allylbromide (0.9 ml, 0.01 mole) in ethyl alcohol (20 ml) was heated on water bath for 3 hours. The reaction mixture was poured onto ice water (200 ml) and stirred for 30 minutes. The obtained solid was filtered and crystallized from absolute ethanol. Yield: 80%; mp80-82°C; IR (KBr, ν, cm-1):3040 (C-H aromatic), 2941(C-H aliphatic), 1640 (C=O amidic). 13C NMR (75 MHz, [D6] DMSO): δ= 24.2, 29.2, 36.5, 41, 118.5, 123.8, 126.3, 126.7, 129.9, 130.4, 130.5, 130.5, 131.0, 131.1, 132.3, 133, 134.5, 136.4, 153.5, 158.2, 163.4. MS (m/z): 438 (M+, 0.35%), 440 (M+2, 0.16%) and base beak at 162 (100%). Anal.Calcd. For C22H19ClN4O2S: C, 60.20; H, 4.36; N, 12.76. Found: C, 60.60; H, 4.48 N, 13.10.2.1.17. Ethyl-5-{[3-(4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl)Propyl]}-[1, 3, 4]-Oxadiazol-2-yl Carbonothioate (8d)Equimolar quantity of the potassium salt (7) (4.36 gm, 0.01 mole) and andethylchloroformate (1.00 ml, 0.01 mole) in DMF (20ml) was heated on a water bath for 2 hours. After cooling to the r. t., the reaction mixture was added to cold water (200ml) with continuous stirring for 30 minutes. The white precipitated product was filtered, washed with water and crystallized from absolute ethanol.Yield: 80%; mp90-91°C; IR (KBr, ν, cm-1):3059 (C-H aromatic), 2952 (C-H aliphatic), 1725 (C=O ester) and 1651 (C=O phthalazine ring). 13C NMR (75 MHz, [D6] DMSO): δ= 13.5, 24.2, 29.3, 41, 63.5, 123.8, 126.3, 127.6, 129.9, 130.4, 130.7, 130.7, 131.0, 131.2, 132.4, 133, 134.4, 136.7, 153.3, 158.2, 158.3, 163.5. MS (m/z): 470 (M+, 0.06%), 472 (M+2, 0.04%) and base beak at 256 (100 %). Anal.Calcd. For C22H19ClN4O4S: C, 56.11; H, 4.07; N, 11.90. Found: C, 56.56; H, 4.38 N, 12.08.2.1.18. 2-[(1-oxo-4--(4-Chlorophenylphthalazin-2(1H)-yl)]-N-(Aryl) acetamide and 3-[(1-oxo-4-(-(4-Chlorophenylphthalazin-2(1H)-yl)]-N-(Aryl)propanamide derivatives (9a-f)General procedureA mixture of 4-(4-chlorophenyl)-phenyl-1(2H)- phthalazinone (2) (2.22 gm, 0.01 mole) and anhydrous K2CO3 (2.75 gm, 0.02 mole) in DMF (30 mL) was heated at 120°C for about one hour, then (0.01 mol) of 2-chloro-N-aryl acetamide or 3-chloro-N-aryl propionamide (prepared according to the reported procedure) [31] and (250 mg) of KI were added to the reaction mixture which allowed to more heating and stirring at 120°C for 7 hours. The reaction mixture was cooled to room temperature then poured on 250 gm of crushed ice. After that precipitate was collected by filtration, washed with ethanol (3 x 10 mL), dried over anhydrous sodium sulfate and crystallized form absolute ethanol.2.1.19. 2-[4-[4-Chlorophenyl]-1-Oxophthalazin-2(1H)-yl]-N-(5-Chloropyridin-2-yl) acetamide(9a)Yield: 79%; mp220-223°C; IR (KBr, ν, cm-1): 3231 (NH), 3045 (C-H aromatic), 1645 (C=O) and 1576 (C=N).1HNMR (300MHz, CDCl3): δ = 1.8 (s, 2H, -CH2CO-), 5.16 (s, 1H, -NCH-), 7.2-7.8 (m, 11 , aromatic protons) and 9.04 (s, 1H , NH). 13C NMR (75 MHz, [D6] DMSO): δ = 55.5, 116.7, 123.8, 126.3, 127.5, 129.0, 130, 130.5, 130.8, 130.8, 131, 131.2, 132.4, 134.9, 136.8, 137.5, 149.3, 150.5, 158.3, 168.4. MS (m/z): 429 (M+4, 4.51%), 427 (M+2, 12.62%), 425 (M+, 4.61%), and base beak at 269 (100%). Anal.Calcd. For C21H14Cl2N4O2: C, 59.31; H, 3.32; N, 13.17. Found: C, 59.08; H, 3.63; N, 13.43.2.1.20. 2-[4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl]-N-(2, 6-Dichlorophenyl) acetamide (9b)Yield: 83%; mp300-302°C; IR (KBr, ν, cm-1): 3189 (NH), 3031(C-Haromatic), 1662 (C=O) and 1535 (C=N).13C NMR (75 MHz, [D6] DMSO): δ= 55.5, 123.7,126.1, 126.3, 127.5, 128.3, 128.3, 129.0, 130.3, 130.5, 130.5,131.0, 131.3, 132.0, 132.3, 133.5, 133.5, 134.8, 136.8, 158.3, 168.8. MS (m/z):461 (M+4, 1.06%), 459 (M+2, 1.06%), 457 (M+, 0.62%), and base beak at 269 (100 %). Anal.Calcd. for C22H14Cl3N3O2: C, 57.60; H, 3.08; N, 9.16. Found: C, 57.92; H, 3.19; N, 9.50.2.1.21. 2-[4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl]-N-(2,6-Dimethylphenyl) acetamide(9c)Yield: 89%; mp 290-292°C; IR (KBr, ν, cm-1): 3230 (NH), 3040 (C-Haromatic), 1657 (C=O) and 1541 (C=N). 1HNMR (300MHz, CDCl3): δ = 2.15 (s, 6H, 2-CH3), 5.03 (s, 2H, -NCH2), 7.1-8.4 (m, 11H, aromatic protons), 9.67 (s, 1H, NH). Anal.Calcd. For C24H20ClN3O2: C, 68.98; H, 4.82; N, 10.06. Found: C, 69.40; H, 4.98; N, 10.15.2.1.22. 3-[4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl]-N-(2,6-Dichlorophenyl) Propanamide(9d)Yield: 80%; mp225-227°C;IR (KBr, ν, cm-1):3163 (NH), 3015 (C-Haromatic), 2960 (C-H aliphatic), 1661 (C=O), 1531 (C=N). 1HNMR (300MHz, CDCl3):δ = 3.1 (t, 2H, -CH2CH2CO-), 4.7 (t, 2H, -NCH2CH2-), 7.1-8.5 (m, 11H, aromatic protons) and 10.7 (s, 1H, NH). 13C NMR (75 MHz, [D6] DMSO): δ= 30.2, 47.6, 123.7, 126.2, 126.3, 127.5, 128.3, 128.3, 129.0, 130.5, 130.8, 130.8,131, 131.2, 132.0, 132.5, 133.5, 133.5, 134.8, 136.8, 158.2, 173.2. Anal.Calcd. For C23H16Cl3N3O2: C, 58.43; H, 3.41; N, 8.89. Found: C, 58.64; H, 3.80; N, 9.10.2.1.23. 3-[4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl]-N-(2,6-Dimethylphenyl) Propanamide (9e)Yield: 86%; mp205-207°C; IR (KBr, ν, cm-1):3291 (NH), 3019 (C-Haromatic), 2937 (C-H aliphatic), 1656 (C=O), 1510 (C=N). MS (m/z): 434 (M+2, 2.28%), 432 (M+1, 6.53%), and base beak at 257.20 (100%). Anal.Calcd. For C25H22ClN3O2: C, 69.62; H, 5.13; N, 9.73. Found: C, 70.10; H, 5.37; N, 9.86.2.1.24. 3-[4-(4-Chlorophenyl)-1-Oxophthalazin-2(1H)-yl]-N-(Thiazol-2-yl) propanamide (9f) Yield: 85%; mp220-222°C; IR (KBr, ν, cm-1): 3280 (NH), 3030 (C-Haromatic), 2950 (C-H aliphatic), 1650 (C=O), 1510 (C=N). 13C NMR (75 MHz, [D6] DMSO): δ= 30.2, 47.5, 112.2, 123.8, 126.3, 127.4, 129.0, 130.4, 130.5, 130.5, 131.0, 131.2, 132.2, 132.3, 132.7, 134.8, 136.7, 158.2, 162.8, 173.9.MS (m/z): 405 (M+, 5.15%), 407 (M+2, 2.6), 223 (98.59%), 182 (56.49%), 184 (17.21%), base beak at 164 (100%), 127 (96.8%) and 129 (31.47%). Anal.Calcd. for C20H15ClN4O2S: C, 58.46; H, 3.68; N, 13.64. Found: C, 58.83; H, 3.98; N, 14.02.
3. Results and Discussion
- The synthetic pathways adopted to obtain the newly synthesized compound 3-9 that was prepared by simple substitution depending on availability of starting material 2 as a good precursor to synthesis of our targeted compounds 3-9. The strategy and aim of this work depend on preparation of starting phthalazinone derivative 3 that obtained and characterized from IR by disappearance of NH stretching and 1HNMR data revealed appearance of aliphatic protons at 1.2-4.3 ppm. The appearance of two protons at δ 4.3ppm and another at δ 8.5ppm which belong the NH2 and NH, both exchangeable with D2O, support the proposed structure 4 as that obtained from the reaction of 3 with hydrazine hydrate. Condensation of 4 with different aldehydes affords Schiff's bases 5a-d, the structures of which was supported by the disappearance of NH2 protons and appearance of azomethene (N=CH) proton at 8.04-8.10ppm. Treatment of 4 with carbon disulfide and potassium hydroxide in refluxing ethanol afforded compound 6 that was then treated with alcoholic potassium hydroxide to produce compound 7. The reaction of 7 with different alkyl halides gave compounds 8a-d that contain anoxadiazolyl ring, the structures of which are substantiated by spectral where two new 13C NMR data signals appear around 153ppm and 163 ppm correlating to the ring system. In addition, the physical data of the produced compounds confirm the production of this series of compounds. Finally, when the keyprecursor 2 was treated with chloroarylacetamide or chloroarylprpionamide it produce compounds 9a-f in which IR spectra confirm the produced compounds in addition to elemental analysis and proton and 13C NMR data. From the previous mentioned discussion, it was clear that our synthetic strategies adopted to obtain the newly synthesized compounds 3-9 using different reagents, were very efficient. In one or two steps, a highly functionalized substituent or heterocyclic ring was added to the molecule that afforded the desired derivatives.
3.1. Molecular Modeling
- The molecular modelling study [32] which led to the three-dimensional (3-D) pharmacophore model for the binding of non-competitive AMPA receptor antagonists in order to map common structural features of highly active compounds, was used by our group in this work. We found that the compound 4-(4-aminophenyl)–6–methoxy–1– methyl–N-propylphthalazine-2(1H)-carboxamide (3 series, Figure 1) mapped well onto the four chemical functionalities of the pharmacophore model: particularly, the carbonyl oxygen of the amido group occupied the hydrogen bond acceptor region, the C-4 phenyl ring was disposed over the aromatic region, whereas the C-1 methyl group and the fused benzene ring of the phthalazine nucleus overlapped with the two hydrophobic features [32]. Taking into consideration the alignment of 4-(4-aminophenyl)–6–methoxy–1–methyl– Npropylphthalazine-2(1H)-carboxamide onto the reported pharmacophore model, we planned to synthesis a new phthalazinone derivative (3, 4, 5a-h, 6-8a-d and 9a-f) that include and meet the structural and molecular requirements. We decided to keep the 4-phenyl substituent and carbonyl oxygen of ester 3 or hydrazido 5a-h or amido 9a-f groups to meet the aromatic region and the hydrogen bond acceptor features described in the 3D model, respectively. Furthermore and depending on Structure-Activity Relationship (SAR) studies [34] which revealed that the increase of lipophilicity is favorable to pharmacological profile; we chose to include different hydrophobic groups on the N-2 side chain in order to meet the hydrophobic feature improving the chance of obtaining active molecules.
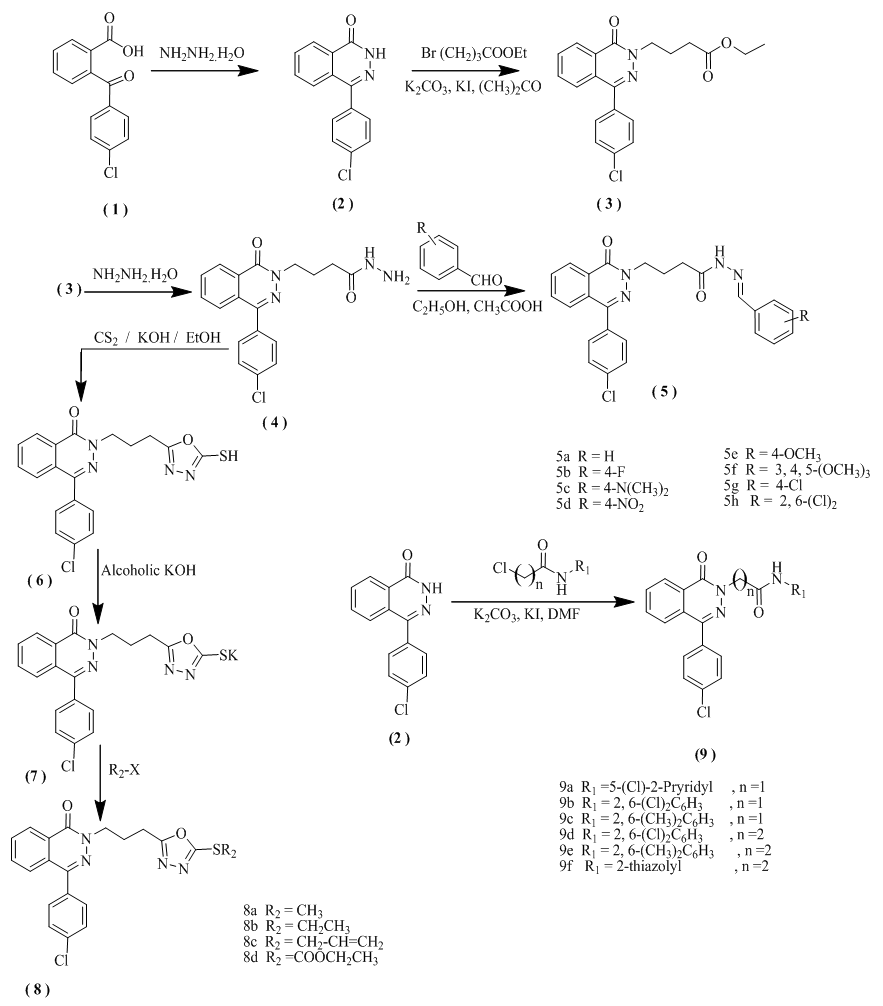 | Scheme 1. Preparation of Phthalazinone Derivatives 3-9 |
3.2. Bioactivity Study
- The mice used were swiss albino adult male mice, weighing between 20 and 25 g, were used as experimental animals. They were obtained from an animal facility (Animal house, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Al-Azhar University). Mice were housed in stainless steel wire-floored cages without any stressful stimuli. Animals were kept under well-ventilated conditions at room temperature (25-30°C). They were fed standard laboratory chow (El Nasr Co., Abou-Zabal, Egypt) and allowed to acclimatize with free access to food and water for 24 hours period before being tested. Albino mice were randomly arranged in groups, each of six animals. Pentylenetetrazole (PTZ, Sigma) was used as convulsive agent and phenobarbitone sodium (Alex Co., Egypt) was used as a reference drug. The Tested compounds were dissolved in DMSO and orally administered in a dose regimen ranging from 200-800 mg/kg animal weight, using the same dosing volume of 0.2 ml per 20 g. Pentylenetetrazole was dissolved in normal saline in 2% concentration and was given intraprotenially in a dose of 60 mg/kg body weight (dose that could induce convulsions in at least 80% of the animals without death during the following 24 hours). Phenobarbitone sodium was dissolved in normal saline in 2% concentration and it was intraprotenially given in doses of 6.25, 12.5 and 25 mg/kg using the same dosing volume. All drugs were freshly prepared to the desired concentration just before use. Groups of six mice were administered the graded doses of the test compounds and phenobarbitone sodium orally. Control animals received an equal volume of saline (10 ml/kg). After one hour, the animals were subcutaneously injected with the convulsive dose of pentylenetetrazole (60 mg/kg). The criterion of anticonvulsant activity is complete protection against convulsions of any kind. Observations were made at least 60 minutes after the administration of pentylenetetrazole. Doses that gave full protection against the induced convulsions and that which exhibited 50% protection in addition to the relative potencies of the test compounds to phenobarbitone sodium were recorded.
3.3. Biological Evaluation
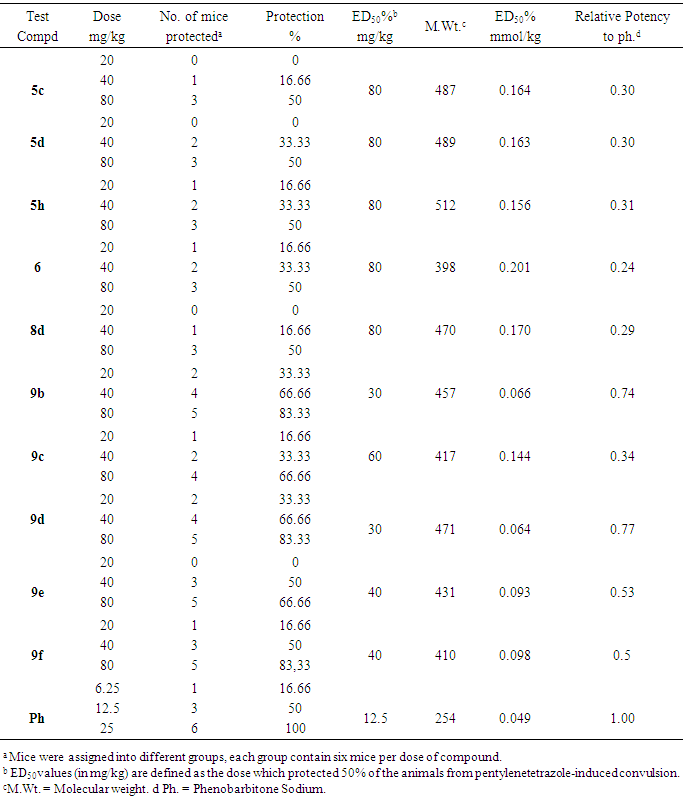
- In this investigation, the synthesized compounds were screened for their anticonvulsant activity by determining their ability to protect experimental animals from pentylenetetrazole-induced convulsions according to the protocol reported by H. Gerhard Vogel [35]. The results were compared with phenobarbitone sodium as a standard anticonvulsant. The percentage protection per each dose and the ED50 of each compound (in mg/kg and in mM) were calculated and presented in Table 1. Finally, the relative potencies of the tested compounds to phenobarbitone sodium was calculated and used for comparison as shown in Table 1. All the target compounds were subjected to docking study together with the internal 4-(4-aminophenyl)-6-methoxy- 1-methyl-N-propylphthalazine-2-(1H)-carboxamideas a reference molecule [36] to explore their calculated binding modes with AMPA-R. The results are showed in Figures 2-4 & Table 2.
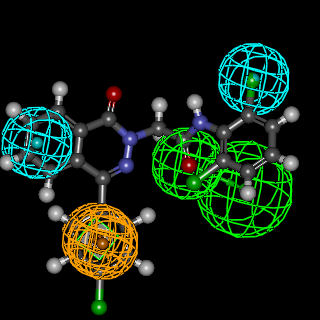 | Figure 2. Mapping of AMPAR antagonist hypothesis and compound 9b |
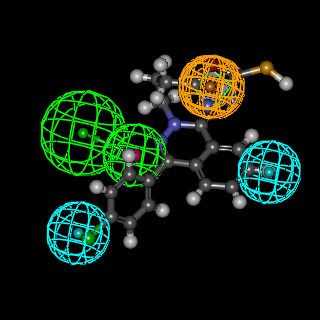 | Figure 3. Mapping of AMPAR antagonist hypothesis and compound 6 |
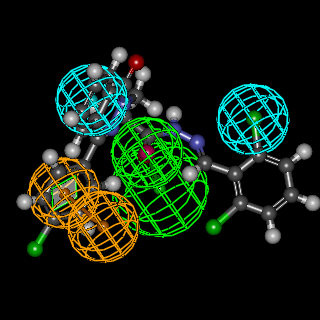 | Figure 4. Mapping of AMPAR antagonist hypothesis and compound 5h |
|
|

4. Conclusions
- In summary, we have designed 22novel phthalazinones as AMPA-R antagonists for the management of convulsion. The target molecules were synthesized from the key intermediate 4-(4-chlorophenyl)-1(2H)-phthalazinone (2). In general, amide derivatives showed better fitting than the benzylidene derivatives. Compound 9b showed the highest fit value because it was perfectly matched to all features in the pharmacophore model. Compound 6 showed the lowest fit value because it was unable to map one of the two hydrophobic features. Replacement of the phenyl ring in the side chain of amide derivatives by other structures led to a decrease in the fitting value. The length of the amido spacer between the phenyl or heterocyclic ring and the phthalazinone moiety is not a key parameter that affects the fitting efficiency of the compounds to the pharmacophore, i.e., elongating the alkylamido chain from acetamido to propanamido did not lead to a notable change in the fitting values. The substitution pattern on the phenyl moiety is a crucial element that accounts for the relationship between structure properties and AMPAR antagonistic effect. Theortho substitution on the phenyl ring of the side chain exhibited higher activities than meta or para substitution. The o-chloro substituent on the phenyl ring of the side chain was highly favorable to AMPAR antagonistic effect, as the o-chlorophenylperfectly matched the hydrophobic system in the pharmacophores model. The presence of the p-nitro group (strong electron withdrawing group), sharply decreases the fitting value. Compound 3 with ester function group showed high fitting value. The efforts here were directed towards the phthalazinones as the biologically-determent part. Compound 6c which showed the highest fit value exhibited the most potent anticonvulsant activity where it incorporated the favorable o-chloro substitution on the phenyl ring of the N-side chain. Compound 4, which showed the lowest fit value, exhibited the least anticonvulsant activity due to the lack of a hydrophobic group required to meet the hydrophobic features. Compound 5b, which showed the highest fit value of the benzylidene series, exhibited the most potent anticonvulsant activity in this series due to o-chloro substitution. Compound 3, which showed a high fit value, exhibited the second most potent anticonvulsant activity probably due to better pharmacokinetic character. This result is compatible with the reported SAR studies [23] which have confirmed the importance of 2 or 2 and 6 substitutions on N-phenyl rings by small, lipophilic groups for anticonvulsant activity.
ACKNOWLEDGMENTS
- The authors would like to express their sincere thanks to Dr. Ahmad Mansour, Pharmacology Department, Faculty of Pharmacy Al-Azher University, Cairo, Egypt, for carrying out the anticonvulsant activity for testing compound. And I would like to thanks all members of Pharmacology Department, Faculty of Pharmacy Al-Azher University, Cairo, Egypt.