-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2016; 6(1): 8-16
doi:10.5923/j.ajoc.20160601.02

Synthesis and Stability of 1,1-Diphenyl-1H-azulenium Cation
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLMitsunori Oda 1, Nobue Nakajima 2, Yoshimitsu Kumai 1, Akira Ohta 1, Ryuta Miyatake 3, Shigeyasu Kuroda 2
1Department of Chemistry, Faculty of Science, Shinshu University, Nagano, Japan
2Deptartment of Applied Chemistry, Graduate School of Science and Engineering, University of Toyama, Toyama, Japan
3Centre for Environmental Conservation and Research Safety, University of Toyama, Toyama, Japan
Correspondence to: Mitsunori Oda , Department of Chemistry, Faculty of Science, Shinshu University, Nagano, Japan.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In three steps by aldol condensation with benzaldehyde, conjugate addition of lithium diphenylcuprate and subsequent oxidation, 1-acetylcyclohepta-1,3,5-triene (10) was transformed into 1-(3,3-diphenylacryloly) -cyclohepta-1,3,5-triene (11). The title cation 9 was synthesized from 11 by a sequence involving Nazarov cyclization, Shapiro reaction and final hydride abstraction with trityl perchlorate. The pKR+ value was determined to be 6.4, which is less than that of the analogous fluorenyl cation 8. Upon heating 9 rearranges to yield 1,2-diphenylazulene (22), quantitatively.
Keywords: Azulene, Carbocation, Nazarov cyclization, Shapiro reaction, pKR+ value
Cite this paper: Mitsunori Oda , Nobue Nakajima , Yoshimitsu Kumai , Akira Ohta , Ryuta Miyatake , Shigeyasu Kuroda , Synthesis and Stability of 1,1-Diphenyl-1H-azulenium Cation, American Journal of Organic Chemistry, Vol. 6 No. 1, 2016, pp. 8-16. doi: 10.5923/j.ajoc.20160601.02.
Article Outline
1. Introduction
- We have investigated synthesis and stability of various 1,1-disubstituted 1H-azulenium cations, 1–8, in order to evaluate an electronic effect of the substituent at the C–1 position of the azulenyl skeleton on its stability (Fig. 1). [1-6] It has been found that cations 1–7 with alkyl and cycloalkyl groups at the position showed a range of pKR+ values, 8.6–10.4, indicating that the cations are stabilized effectively by electron-donating nature of those groups. On the other hand, the pKR+ value of cation 8, having a fluorenyl moiety at the position, was found to be 7.8. The relative instablilty of 8 compared with those of 1–7 can be ascribed to weak electron-withdrawing nature of sp2-hybridized carbon atoms of the fluorenyl group. Noteworthily, cation 8 shows an intramolecular charge-transfer (CT) excitation from the fluorenyl part to the azulenium ion part in its UV-vis absorption spectrum. [6] This phenomenon surprised us, since the planes of the two parts are expected to be perpendicular to each other based on the structure optimized by DFT calculations. In order to evaluate further an effect of a simple phenyl group on stability of the azulenium cation and an intramolecular CT, synthesis of the title ion, 1,1-diphenyl-1H-azulenium perchlorate (9), was contrived. Herein, we disclose a result of its synthesis, stability and spectroscopic properties.
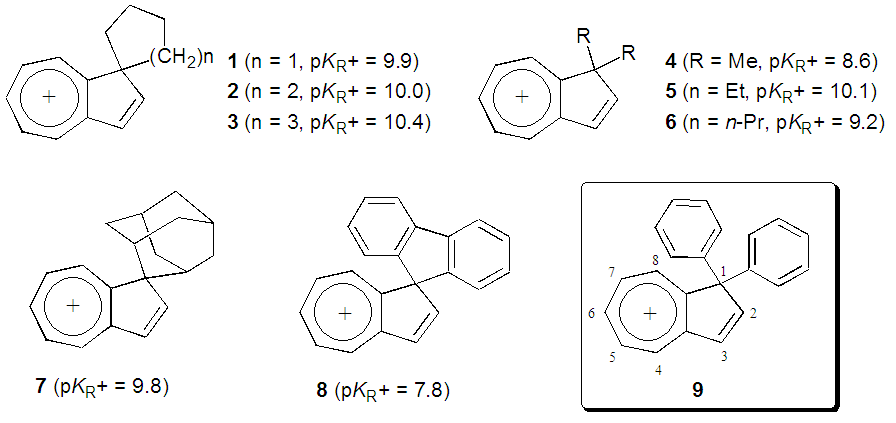 | Figure 1. Various 1,1-disubstituted 1H-azulenium cations and their pKR+ values |
2. Results and Discussion
- An original synthetic plan for 9 from 1-acetylcyclohepta- 1,3,5-triene (10) is shown in Scheme 1, which is based on our previous syntheses, [7–9] involving aldol condensation of 10 with benzophenone, Nazarov cyclization [10] of 11, transformation from 12 to 13, and final hydride abstraction.
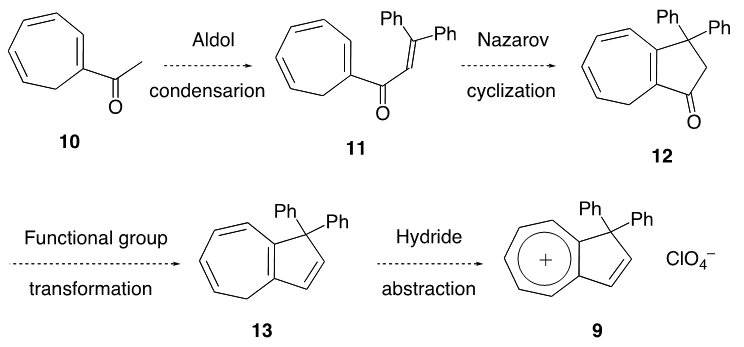 | Scheme 1. A synthetic plan from 10 to 9 at the beginning |
 | Scheme 2. An attempted synthetic approach from 10 toward 11 |
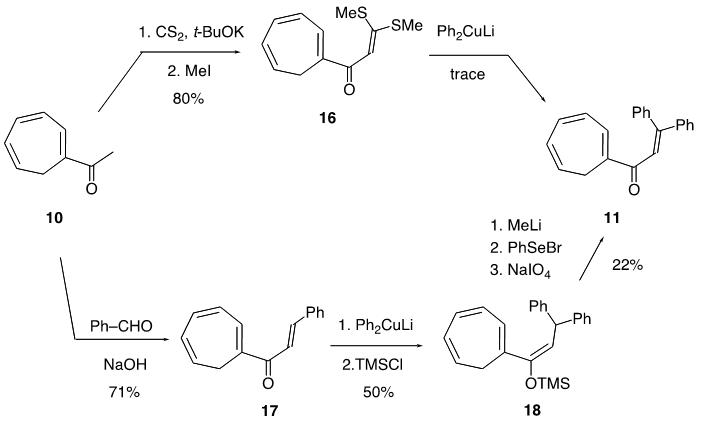 | Scheme 3. Synthetic routes from 10 to 11 |
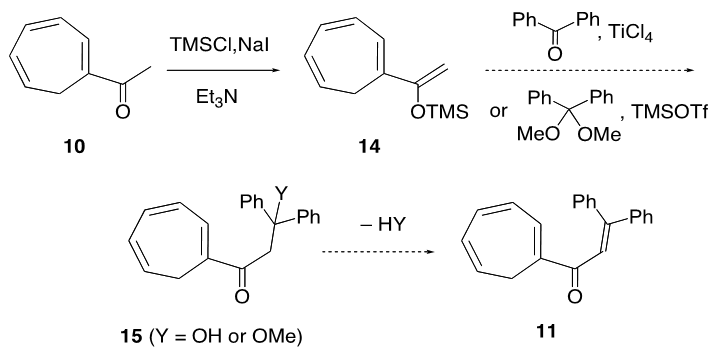
 intermediate provided 11 in 22% yield, accompanied with a trace amount of 1-(3,3-diphenylpropionyl)cyclohepta-1,3,5- triene, which may be derived from hydrolysis of 18. With compound 11 in our hands, its transformation into 9 was carried out by a previously reported three-step sequence (Scheme 4). It is worthy to note that the Nazarov cyclization of 11 gave not only 12 but also its regioisomer 19 as a by-product, as seen in previous results of structurally related substrates. [5, 7] The Shapiro reaction [15] of tosylhydrazone of 12 provided hydrocarbon 14 and the title cation 9 was obtained as a perchlorate salt by hydride abstraction of 14 with trityl perchlorate. Cation 9 was isolated as slightly greenish yellow crystals and its structure was supported by spectroscopic and combustion analyses. Selected assigned NMR signals of 9 with those of 8 [16] are shown in Fig. 2. The proton signals around the azulenyl ring in 9 were observed similar as seen in 4 and 5. We reported that proton signals at the 2 and 8 positions in 8 were observed high-field shifted
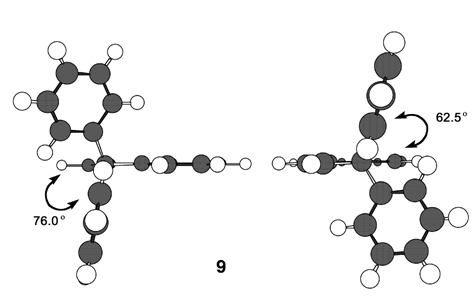
intermediate provided 11 in 22% yield, accompanied with a trace amount of 1-(3,3-diphenylpropionyl)cyclohepta-1,3,5- triene, which may be derived from hydrolysis of 18. With compound 11 in our hands, its transformation into 9 was carried out by a previously reported three-step sequence (Scheme 4). It is worthy to note that the Nazarov cyclization of 11 gave not only 12 but also its regioisomer 19 as a by-product, as seen in previous results of structurally related substrates. [5, 7] The Shapiro reaction [15] of tosylhydrazone of 12 provided hydrocarbon 14 and the title cation 9 was obtained as a perchlorate salt by hydride abstraction of 14 with trityl perchlorate. Cation 9 was isolated as slightly greenish yellow crystals and its structure was supported by spectroscopic and combustion analyses. Selected assigned NMR signals of 9 with those of 8 [16] are shown in Fig. 2. The proton signals around the azulenyl ring in 9 were observed similar as seen in 4 and 5. We reported that proton signals at the 2 and 8 positions in 8 were observed high-field shifted 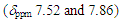 by a shielding effect of the fluorene moiety, supporting the perpendicular relationship between the azulenium ion part and the fluorene ring. The proton shift values of 9 indicate that the arrangement of the phenyl groups at the 1 position in 9 is different from that in 8. [17] The optimized structure of 9 by DFT calculation at B3LYP/6-31G (d) level of theory [18] is shown in Fig. 3, which indeed shows two phenyl rings are connected to the azulenyl ring, not perpendicularly, with angles of 62.5° and 76.0°.
by a shielding effect of the fluorene moiety, supporting the perpendicular relationship between the azulenium ion part and the fluorene ring. The proton shift values of 9 indicate that the arrangement of the phenyl groups at the 1 position in 9 is different from that in 8. [17] The optimized structure of 9 by DFT calculation at B3LYP/6-31G (d) level of theory [18] is shown in Fig. 3, which indeed shows two phenyl rings are connected to the azulenyl ring, not perpendicularly, with angles of 62.5° and 76.0°.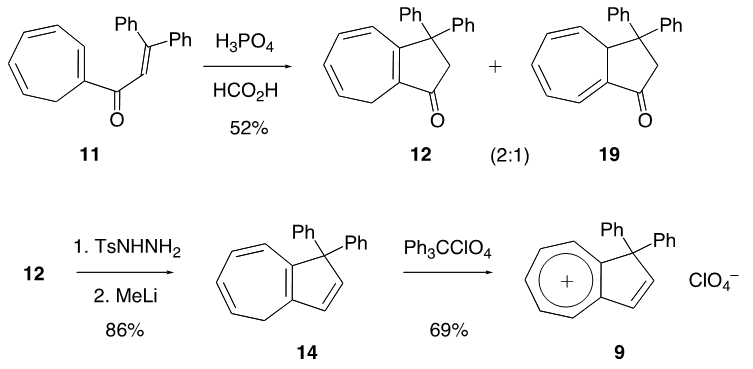 | Scheme 4. Synthesis of 9 from 11 |
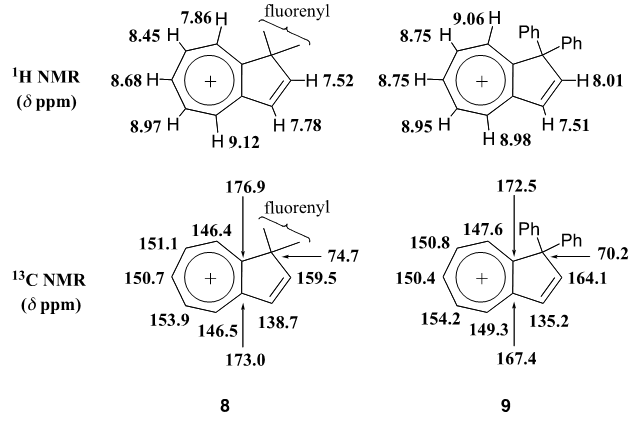 | Figure 2. Assigned 1H- and 13C-NMR signals of the azulenium ion part of 8 an 9 |
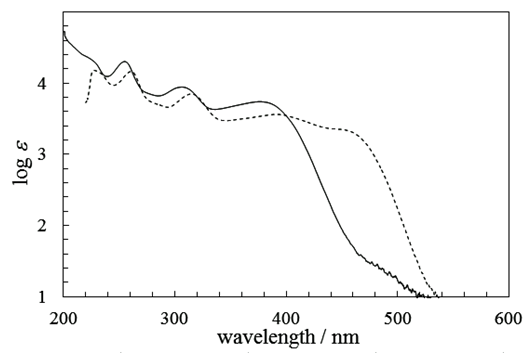 | Figure 3. Optimized structure (Chem3D output) of 9 by DFT calculation at B3LYP/6-31G (d) level of theory |
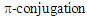 of the azulenium ion part seen in the spectra of 1–7 and, therefore, is thought to be intramolecular CT absorption from the HOMO, whose
of the azulenium ion part seen in the spectra of 1–7 and, therefore, is thought to be intramolecular CT absorption from the HOMO, whose coefficients distribute almost at the phenyl group, to the LUMO, whose
coefficients distribute almost at the phenyl group, to the LUMO, whose  coefficients distribute almost at the azulenium ion part. [19] However, its intensity
coefficients distribute almost at the azulenium ion part. [19] However, its intensity 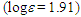 is smaller than that
is smaller than that 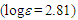 of 8 in CH2Cl2, probably due to different arrangement between the phenyl and fluorenyl rings at the 1 position. The pKR+ value of 9 was determined to be 6.4 by the UV method in 50% aqueous acetonitrile. Thus, the stability of 9 is greater than that of the tropylium ion (3.9), and less by about 1.4 pKR+ units than that of 8, suggesting that electron-withdrawing nature of the phenyl groups in 9 works more effectively than that of the fluorenyl group to destabilize the cationic part. The relative thermodynamical stability of 8 and 9 is reflected in their ability for thermal rearrangement. While 8 did not show any rearrangement at 100°C, 9 undergoes rearrangement above 80°C in acetonitrile to yield 1,2-diphenylazulene (22) [20] quantitatively (Scheme 5).
of 8 in CH2Cl2, probably due to different arrangement between the phenyl and fluorenyl rings at the 1 position. The pKR+ value of 9 was determined to be 6.4 by the UV method in 50% aqueous acetonitrile. Thus, the stability of 9 is greater than that of the tropylium ion (3.9), and less by about 1.4 pKR+ units than that of 8, suggesting that electron-withdrawing nature of the phenyl groups in 9 works more effectively than that of the fluorenyl group to destabilize the cationic part. The relative thermodynamical stability of 8 and 9 is reflected in their ability for thermal rearrangement. While 8 did not show any rearrangement at 100°C, 9 undergoes rearrangement above 80°C in acetonitrile to yield 1,2-diphenylazulene (22) [20] quantitatively (Scheme 5).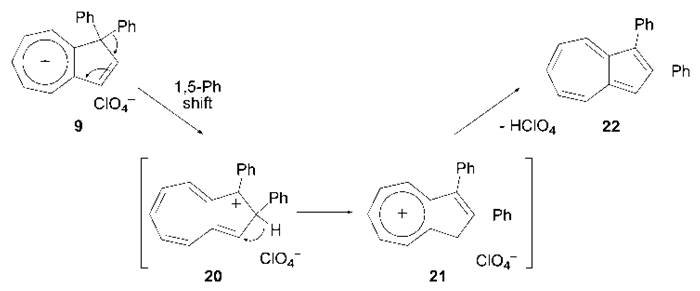 | Figure 4. UV-Vis absorption spectra of cation 9 in acetonitrile (solid line), and dichloromethane (broken line) |
 | Scheme 5. Thermal rearrangement of 9 |
3. Experimental
3.1. General Remarks
- Melting points were measured on a Yanaco MP-3 and are uncorrected. IR spectra were recorded on a PERKIN ELMER Spectrum RX-II spectrometer. 1H- and 13C-NMR spectra were recorded on a
 spectrometer. Chemical shift values of tetramethylsilane
spectrometer. Chemical shift values of tetramethylsilane  for 1H-NMR spectra and CDCl3
for 1H-NMR spectra and CDCl3  for 13C-NMR spectra were used as internal standard. Mass spectra were measured on a JMS-700 mass spectrometer. Column chromatography was performed with Kiesel gel 60F from Merck Co. Benzaldehyde, cyclohepta-1,3,5-triene, acetyl chloride, and tosylhydrazine were purchased from Tokyo Chemical Ind. Chlorotrimethylsilane was purchased from Kanto Chem. and was distilled over CaH2. Ether and tetrahydrofuran (THF) were distilled before use over sodium benzophene ketyl radical under nitrogen atomosphere. Acetonitrile was purchased from Kanto Chem. and was distilled over P2O5. A phenyllithium solution in ether was purchased from Tokyo Chemical Ind. A methyllithium solution in ether and phenylselenyl bromide were purchased from Aldrich Co. Compound 10 was obtained by acetylation of cyclohepta-1,3,5-triene with acetyl chloride and zinc chloride in dichloromethane. [21] Trityl perchlorate was prepared according to a method of Dauben et al. and was used after purification by recrystallization from dichloromethane-hexane. [22]
for 13C-NMR spectra were used as internal standard. Mass spectra were measured on a JMS-700 mass spectrometer. Column chromatography was performed with Kiesel gel 60F from Merck Co. Benzaldehyde, cyclohepta-1,3,5-triene, acetyl chloride, and tosylhydrazine were purchased from Tokyo Chemical Ind. Chlorotrimethylsilane was purchased from Kanto Chem. and was distilled over CaH2. Ether and tetrahydrofuran (THF) were distilled before use over sodium benzophene ketyl radical under nitrogen atomosphere. Acetonitrile was purchased from Kanto Chem. and was distilled over P2O5. A phenyllithium solution in ether was purchased from Tokyo Chemical Ind. A methyllithium solution in ether and phenylselenyl bromide were purchased from Aldrich Co. Compound 10 was obtained by acetylation of cyclohepta-1,3,5-triene with acetyl chloride and zinc chloride in dichloromethane. [21] Trityl perchlorate was prepared according to a method of Dauben et al. and was used after purification by recrystallization from dichloromethane-hexane. [22]3.2. 1-Cinnamoylcyclohepta-1,3,5-Triene (17)
- A solution of 10.1 g (7.54 mmol) of 10 in 60 mL of methanol was added slowly to a solution of benzaldehyde (6.06 g, 5.72 mmol) and sodium methoxide (12.3 g, 228 mmol) in 60 mL of methanol. The mixture was stirred at room temperature for 21 h, and then was poured into a 1M HCl solution and extracted with chloroform (50 mL x 4). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. The solvent was removed under vacuum and the residue was purified by silica gel chromatography (hexane/toluene = 3/7) to give 9.01 g (71% yield) of 17 as a pale yellow oil.17: IR (liq. film)
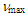 = 1597 (s), 1575 (s), 1305 (s), 1200 (s), 1167 (s), 761 (s), 715 (s) cm–1; 1H NMR (CDCl3, 400 MHz)
= 1597 (s), 1575 (s), 1305 (s), 1200 (s), 1167 (s), 761 (s), 715 (s) cm–1; 1H NMR (CDCl3, 400 MHz) 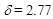 (d, J = 7.0 Hz, 2H), 5.62 (dt, J = 9.6, 7.0 Hz, 1H), 6.29 (dd, J = 9.6, 5.6 Hz ,1H), 6.73 (dd, J = 11.2, 5.6 Hz, 1H), 6.86 (dd, J = 11.2, 5.6 Hz, 1H), 7.19 (d, J = 5.6 Hz, 1H), 7.35 (J = 15.6 Hz, 1H), 7.40 (m, 3H), 7.57 (m, 2H), 7.65 (d, J = 15.6 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz)
(d, J = 7.0 Hz, 2H), 5.62 (dt, J = 9.6, 7.0 Hz, 1H), 6.29 (dd, J = 9.6, 5.6 Hz ,1H), 6.73 (dd, J = 11.2, 5.6 Hz, 1H), 6.86 (dd, J = 11.2, 5.6 Hz, 1H), 7.19 (d, J = 5.6 Hz, 1H), 7.35 (J = 15.6 Hz, 1H), 7.40 (m, 3H), 7.57 (m, 2H), 7.65 (d, J = 15.6 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz) 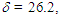 121.8, 125.6, 127.1, 128.2, 128.8, 129.2, 130.2, 132.2, 132.7, 135.0, 135.9, 143.3, 189.3 ppm; UV (CH3OH)
121.8, 125.6, 127.1, 128.2, 128.8, 129.2, 130.2, 132.2, 132.7, 135.0, 135.9, 143.3, 189.3 ppm; UV (CH3OH) 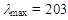
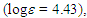 227 (4.09), 321 (4.26) nm; MS m/z (rel. int) 222 (M+, 55 %), 221 (29), 207 (29), 131 (100), 103 (31), 77 (56). HRMS calcd for C16H14O 222.1045, found 222.1050.
227 (4.09), 321 (4.26) nm; MS m/z (rel. int) 222 (M+, 55 %), 221 (29), 207 (29), 131 (100), 103 (31), 77 (56). HRMS calcd for C16H14O 222.1045, found 222.1050.3.3. 1-(3,3-Diphenylacryloyl)Cyclohepta-1,3,5-Triene (11) from 17
- A solution of 0.4 M phenyllithium solution in ether (50 mL, 20 mmol) was added slowly to a suspension of cupric iodide (1.90 g, 10.0 mmol) in 15 mL of ether at –20°C. To a resulted greenish solution of lithium diphenylcuprate was added a solution of 11 (2.23g, 10.0 mmol) in 30 mL of ether at the same temperature. After being stirred at room temperature for 1 h, the reaction mixture was quenched by adding 2.54 mL (20.0 mmol) of chlorotrimethylsilane and then stirred further for 4 h. The resulted reaction mixture was poured into water and solids were removed by passing through a Celite pad. The filtrate was extracted with ether (50mL x 3) and the combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under vacuum and the residue was passed quickly through silica gel short column (hexane/ether = 95/5) to give 1.82 g (50% yield) of crude 18 as a pale yellow oil. Without further purification, this crude sample was used in a next step.18: IR (liq. film)
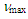 = 3025 (s), 1493 (s), 1252 (s), 1071 (s), 1041 (s), 1028 (s), 895 (s), 845 (s), 757 (s), 741 (s), 699 (s) cm–1; 1H NMR of the major isomer (CDCl3, 400 MHz)
= 3025 (s), 1493 (s), 1252 (s), 1071 (s), 1041 (s), 1028 (s), 895 (s), 845 (s), 757 (s), 741 (s), 699 (s) cm–1; 1H NMR of the major isomer (CDCl3, 400 MHz) 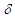 = 0.07 (s, 9H), 2.55 (d, J = 7.2 Hz, 2H), 5.15 (d, J = 9.9 Hz, 1H), 5.40 (dt, J = 9.2, 7.2 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 6.19 (dd, J = 9.2, 5.3 Hz, 1H), 6.44 (d, J = 5.9 Hz, 1H), 6.54 (dd, J = 11.0, 5.3 Hz, 1H), 6.60 (dd, J = 11.0, 5.9 Hz, 1H), 7.15–7.30 (m, 10H) ppm; MS m/z (rel. int) 372 (M+, 100), 371 (42), 357 (24), 295 (11), 282 (14), 281 (23), 205 (14), 191 (14), 179 (20), 178 (19), 167 (16), 165 (29), 152 (11), 115 (10). HRMS Calcd for C25H28OSi 372.1884, found 372.1882.To a solution of crude 18 (186 mg, 0.500 mmol) in 10 mL of THF at 0°C was added a 1.25 M methyllithium solution in ether (0.44 mL, 0.55 mmol), followed by 118 mg (0.500 mmol) of phenylselenyl bromide with 5 mL of THF. The reaction mixture was stirred at room temperature for 4 h, and then was poured into water and extracted with ether (40 mL x 3). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. After being dried over MgSO4, the solvent was removed under vacuum. The residue was dissolved in 5 mL of methanol. To this solution was added 107 mg (0.500 mmol) of NaIO4 and 0.5 mL of water. The mixture was refluxed on an oil bath for 3 h, and then was poured into water and extracted with chloroform (20 mL x 3). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. The solvent was removed under vacuum and the residue was purified by silica gel chromatography (AcOEt/hexane = 18/82) to give 33 mg (22% yield) of 11 as a pale yellow oil, accompanied with 4 mg (3% yield) of 1-(3,3-diphenylpropionyl)cyclohepta-1,3,5-triene as creamy white solids.11: IR (liq. film)
= 0.07 (s, 9H), 2.55 (d, J = 7.2 Hz, 2H), 5.15 (d, J = 9.9 Hz, 1H), 5.40 (dt, J = 9.2, 7.2 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 6.19 (dd, J = 9.2, 5.3 Hz, 1H), 6.44 (d, J = 5.9 Hz, 1H), 6.54 (dd, J = 11.0, 5.3 Hz, 1H), 6.60 (dd, J = 11.0, 5.9 Hz, 1H), 7.15–7.30 (m, 10H) ppm; MS m/z (rel. int) 372 (M+, 100), 371 (42), 357 (24), 295 (11), 282 (14), 281 (23), 205 (14), 191 (14), 179 (20), 178 (19), 167 (16), 165 (29), 152 (11), 115 (10). HRMS Calcd for C25H28OSi 372.1884, found 372.1882.To a solution of crude 18 (186 mg, 0.500 mmol) in 10 mL of THF at 0°C was added a 1.25 M methyllithium solution in ether (0.44 mL, 0.55 mmol), followed by 118 mg (0.500 mmol) of phenylselenyl bromide with 5 mL of THF. The reaction mixture was stirred at room temperature for 4 h, and then was poured into water and extracted with ether (40 mL x 3). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. After being dried over MgSO4, the solvent was removed under vacuum. The residue was dissolved in 5 mL of methanol. To this solution was added 107 mg (0.500 mmol) of NaIO4 and 0.5 mL of water. The mixture was refluxed on an oil bath for 3 h, and then was poured into water and extracted with chloroform (20 mL x 3). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. The solvent was removed under vacuum and the residue was purified by silica gel chromatography (AcOEt/hexane = 18/82) to give 33 mg (22% yield) of 11 as a pale yellow oil, accompanied with 4 mg (3% yield) of 1-(3,3-diphenylpropionyl)cyclohepta-1,3,5-triene as creamy white solids.11: IR (liq. film) 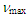 = 1644 (s), 1603 (s), 1265 (s), 1200 (s), 1167 (s), 774 (s), 715 (s), 698 (s) cm–1.1H NMR (CDCl3, 400 MHz) d = 2.56 (d, J = 7.1 Hz, 2H), 5.48 (dt, J = 9.3, 7.1 Hz, 1H), 6.18 (dd, J = 9.3, 5.6 Hz, 1H), 6.57 (dd, J = 11.2, 6.1 Hz, 1H), 6.75 (dd, J = 11.1, 5.5 Hz, 1H), 6.83 (s, 1H), 7.08 (d, J = 5.9 Hz, 1H), 7.12–7.15 (m, 2H), 7.27–7.37 (m, 8H) ppm; 13C NMR (CDCl3, 100 MHz)
= 1644 (s), 1603 (s), 1265 (s), 1200 (s), 1167 (s), 774 (s), 715 (s), 698 (s) cm–1.1H NMR (CDCl3, 400 MHz) d = 2.56 (d, J = 7.1 Hz, 2H), 5.48 (dt, J = 9.3, 7.1 Hz, 1H), 6.18 (dd, J = 9.3, 5.6 Hz, 1H), 6.57 (dd, J = 11.2, 6.1 Hz, 1H), 6.75 (dd, J = 11.1, 5.5 Hz, 1H), 6.83 (s, 1H), 7.08 (d, J = 5.9 Hz, 1H), 7.12–7.15 (m, 2H), 7.27–7.37 (m, 8H) ppm; 13C NMR (CDCl3, 100 MHz) 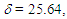 124.90, 125.70, 127.01, 127.83, 128.11, 128.20, 128.37, 128.49, 129.02, 129.63 (2C), 132.96, 133.63, 135.55, 141.26, 152.49, 193.19 ppm; UV (CH3OH)
124.90, 125.70, 127.01, 127.83, 128.11, 128.20, 128.37, 128.49, 129.02, 129.63 (2C), 132.96, 133.63, 135.55, 141.26, 152.49, 193.19 ppm; UV (CH3OH) 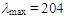
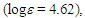 226sh (4.30), 255sh (4.05), 321 (3.99) nm; MS m/z (rel. int) 298 (M+, 100), 297 (21), 221 (11), 207 (51), 179 (42), 178 (90), 177 (12), 176 (12), 167 (14), 165 (12), 152 (14), 133 (11), 105 (20), 91 (20). HRMS Calcd for C22H18O 298.1338, found 298.1335.1-(3,3-Diphenylpropionyl)cyclohepta-1,3,5-triene: M.p. 70–72°C; IR (KBr)
226sh (4.30), 255sh (4.05), 321 (3.99) nm; MS m/z (rel. int) 298 (M+, 100), 297 (21), 221 (11), 207 (51), 179 (42), 178 (90), 177 (12), 176 (12), 167 (14), 165 (12), 152 (14), 133 (11), 105 (20), 91 (20). HRMS Calcd for C22H18O 298.1338, found 298.1335.1-(3,3-Diphenylpropionyl)cyclohepta-1,3,5-triene: M.p. 70–72°C; IR (KBr) 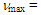 1656 (s), 720 (s), 703 (s) cm-1; 1H NMR (CDCl3, 400 MHz)
1656 (s), 720 (s), 703 (s) cm-1; 1H NMR (CDCl3, 400 MHz) 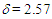 (d, J = 7.1 Hz, 2H), 3.47 (d, J = 7.3 Hz, 2H), 4.70 (t, J = 7.3 Hz, 1H), 5.50 (dt, J = 9.0, 7.1 Hz, 1H), 6.23 (dd, J = 9.4, 5.7 Hz, 1H), 6.66 (dd, J = 11.2, 6.1 Hz, 1H), 6.83 (dd, J = 11.2, 5.6 Hz, 1H), 7.10 (d, J = 6.1 Hz, 1H), 7.16 (tt, J = 7.0, 1.8 Hz, 2H), 7.20–7.28 (m, 8H) ppm; 13C NMR (CDCl3, 100 MHz)
(d, J = 7.1 Hz, 2H), 3.47 (d, J = 7.3 Hz, 2H), 4.70 (t, J = 7.3 Hz, 1H), 5.50 (dt, J = 9.0, 7.1 Hz, 1H), 6.23 (dd, J = 9.4, 5.7 Hz, 1H), 6.66 (dd, J = 11.2, 6.1 Hz, 1H), 6.83 (dd, J = 11.2, 5.6 Hz, 1H), 7.10 (d, J = 6.1 Hz, 1H), 7.16 (tt, J = 7.0, 1.8 Hz, 2H), 7.20–7.28 (m, 8H) ppm; 13C NMR (CDCl3, 100 MHz) 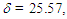 44.28, 46.47, 125.70, 126.31, 127.05, 127.83, 128.49, 129.08, 131.51, 132.02, 135.93, 144.17, 197.49 ppm; UV (CH3OH)
44.28, 46.47, 125.70, 126.31, 127.05, 127.83, 128.49, 129.08, 131.51, 132.02, 135.93, 144.17, 197.49 ppm; UV (CH3OH) 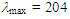
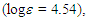 2.18sh (4.39), 271sh (3.70), 294 (3.73) nm; MS m/z (rel. int) 300 (M+, 21), 181 (18), 179 (12), 178 (15), 168 (17), 167 (100), 166 (21), 165 (52), 152 (25), 134 (21), 133 (83), 119 (10), 105 (17), 103 (21), 91 (36). HRMS Calcd for C22H20O 300.1514, found 300.1511. Elemantal analysis; calcd for C22H20O C 87.96, H 6.71%, found C 87.68, H 6.77%.
2.18sh (4.39), 271sh (3.70), 294 (3.73) nm; MS m/z (rel. int) 300 (M+, 21), 181 (18), 179 (12), 178 (15), 168 (17), 167 (100), 166 (21), 165 (52), 152 (25), 134 (21), 133 (83), 119 (10), 105 (17), 103 (21), 91 (36). HRMS Calcd for C22H20O 300.1514, found 300.1511. Elemantal analysis; calcd for C22H20O C 87.96, H 6.71%, found C 87.68, H 6.77%.3.4. 1-[3,3-Bis(Thiomethyl)Acryloyl]Cyclohepta-1,3,5- Triene (16)
- A solution of 1.34 g (10.0 mmol) of 10 in 10 mL of THF was added to a suspension of 2.24 g (20.0 mmol) of t-BuOK in 20 mL of THF, followed by addition of carbon disulfide (0.60 mL, 10.0 mmol), and then iodomethane (1.34 mL, 22.0 mmol). After being stirred at room temperature for 2 hr, the reaction mixture was poured in water and extracted with dichloromehane (30 mL x 3). The combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography (AcOEt/hexane = 4/1) to give 1.90 g (80%) of 16 as yellow needles. 16: M.p. 77–78°C; IR (KBr)
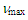 = 1597 (s), 1483 (s), 1472 (s), 1170 (s) cm–1; 1H NMR (CDCl3, 400 MHz)
= 1597 (s), 1483 (s), 1472 (s), 1170 (s) cm–1; 1H NMR (CDCl3, 400 MHz) 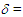 2.46 (s, 6H), 2.70 (d, J = 7.2 Hz, 2H), 5.57 (dt, J = 9.4, 6.8 Hz, 1H), 6.26 (dd, J = 9.4, 5.6 Hz, 1H), 6.55 (s, 1H), 6.68 (dd, J = 11.2, 6.2 Hz, 1H), 6.81 (dd, J = 11.2, 5.6 Hz, 1H), 6.84 (d, J = 6.2 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz)
2.46 (s, 6H), 2.70 (d, J = 7.2 Hz, 2H), 5.57 (dt, J = 9.4, 6.8 Hz, 1H), 6.26 (dd, J = 9.4, 5.6 Hz, 1H), 6.55 (s, 1H), 6.68 (dd, J = 11.2, 6.2 Hz, 1H), 6.81 (dd, J = 11.2, 5.6 Hz, 1H), 6.84 (d, J = 6.2 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz) 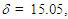 17.27, 26.23, 110.07, 124.95, 126.91, 129.18, 129.24, 133.39, 134.87, 164.09, 185.27 ppm; UV (CH3OH)
17.27, 26.23, 110.07, 124.95, 126.91, 129.18, 129.24, 133.39, 134.87, 164.09, 185.27 ppm; UV (CH3OH) 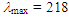
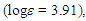 360 (4.27) nm; MS m/z (rel. int) 238 (M+, 25), 208 (10), 191 (28), 175 (29), 161 (50), 144 (73), 133 (38), 118 (41), 91 (100), 77 (15), 75 (38), 65 (42). HRMS Calcd for C12H14OS2 238.0486, found 238.0490. Elemental analysis; calcd for C12H14OS2 C 60.46, H 5.92%, found C 60.78, H 5.95%.
360 (4.27) nm; MS m/z (rel. int) 238 (M+, 25), 208 (10), 191 (28), 175 (29), 161 (50), 144 (73), 133 (38), 118 (41), 91 (100), 77 (15), 75 (38), 65 (42). HRMS Calcd for C12H14OS2 238.0486, found 238.0490. Elemental analysis; calcd for C12H14OS2 C 60.46, H 5.92%, found C 60.78, H 5.95%.3.5. 1-(3,3-Diphenylacryloyl)Cyclohepta-1,3,5-Triene (11) from 16
- A solution of 0.4 M phenyllithium solution in ether (20 mL, 10.0 mmol) was added slowly to a suspension of cupric iodide (952 mg, 5.00 mmol) in 10 mL of ether at –20°C. To a resulted greenish solution of lithium diphenylcuprate was added a solution of 16 (595 mg, 2.50 mmol) in 7 mL of ether at the same temperature. After the reaction mixture was stirred at room temperature for 10 h, the resulted reaction mixture was poured into 0.05M HCl (70 mL) and solids formed were removed by passing through a Celite pad. The filtrate was extracted with ether (50mL x 3) and the combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under vacuum and the residue was purified by silica gel chromatography (AcOEt/hexane = 18/82) to give 22 mg (3% yield) of 11 as a pale yellow oil.
3.6. Nazarov Cyclization of 1-(3,3-Diphenylacryloyl)cyclohepta-1,3,5-Triene (11)
- A solution of 11 (2.37g, 7.95 mmol) in a mixture of phosphoric acid (50 mL) and formic acid (50 mL) was heated on an oil bath at 90°C for 24 h. After being cooled to room temperature, the resulted reaction mixture was poured into water and extracted with ether (80 mL x 3). The combined organic layer was washed with a saturated NaHCO3 aqueous solution and brine. The solvent was removed under vacuum and the residue was purified by silica gel chromatography (AcOEt/hexane = 5/95) to give 903 mg (38% yield) of 12 as yellow solids and 448 mg (14% yield) of 19 as yellow microcrystals.12: M.p. 142–144°C; IR (KBr)
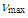 = 1688 (s), 1655 (m), 703 (s) cm–1; 1H NMR (CDCl3, 400 MHz)
= 1688 (s), 1655 (m), 703 (s) cm–1; 1H NMR (CDCl3, 400 MHz) 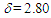 (d, J = 6.5 Hz, 2H), 3.25 (s, 2H), 5.73 (dt, J = 9.9, 6.5 Hz, 1H), 6.20 (dd, J = 9.9, 6.0 Hz, 1H), 6.44 (d, J = 11.5 Hz, 1H), 6.72 (dd, J = 11.5, 6.0 Hz, 1H), 7.11–7.32 ppm; UV (CH3OH)
(d, J = 6.5 Hz, 2H), 3.25 (s, 2H), 5.73 (dt, J = 9.9, 6.5 Hz, 1H), 6.20 (dd, J = 9.9, 6.0 Hz, 1H), 6.44 (d, J = 11.5 Hz, 1H), 6.72 (dd, J = 11.5, 6.0 Hz, 1H), 7.11–7.32 ppm; UV (CH3OH) 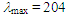
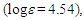 212sh (4.47), 273sh (3.64), 297 (3.72) nm; MS m/z (rel. int) 298 (M+, 73), 256 (42), 255 (23), 239 (16), 221 (28), 220 (29), 219 (15), 207 (15), 193 (16), 192 (29), 191 (50), 189 (33), 179 (45), 178 (100), 176 (20), 165 (58), 152 (25), 118 (17), 115 (35). HRMS calcd for C22H18O 298.1358, found 298.1348. Elemental analysis calcd for C22H18O C, 88.56, H 6.08 %; found C, 88.76, H, 6.19%.19: M.p. 42–44°C; IR (KBr)
212sh (4.47), 273sh (3.64), 297 (3.72) nm; MS m/z (rel. int) 298 (M+, 73), 256 (42), 255 (23), 239 (16), 221 (28), 220 (29), 219 (15), 207 (15), 193 (16), 192 (29), 191 (50), 189 (33), 179 (45), 178 (100), 176 (20), 165 (58), 152 (25), 118 (17), 115 (35). HRMS calcd for C22H18O 298.1358, found 298.1348. Elemental analysis calcd for C22H18O C, 88.56, H 6.08 %; found C, 88.76, H, 6.19%.19: M.p. 42–44°C; IR (KBr) 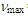 = 1709 (s), 1617 (s), 699 (s) cm–1; 1H NMR (CDCl3,400 MHz)
= 1709 (s), 1617 (s), 699 (s) cm–1; 1H NMR (CDCl3,400 MHz) 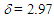 (dm, J = 6.3 Hz, 1H), 2.99 (d, J = 16.4 Hz, 1H), 3.38 (d, J = 16.4 Hz, 1H), 5.12 (dd, J = 9.5, 6.3 Hz, 1H), 6.22 (m, 1H), 6.97 (m, 2H), 7.06 (m, 2H), 7.11–7.14 (m, 2H), 7.20 (tm, J = 7.3 Hz, 2H), 7.24–7.31 (m, 3H), 7.40 (tm, J = 7.6 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz)
(dm, J = 6.3 Hz, 1H), 2.99 (d, J = 16.4 Hz, 1H), 3.38 (d, J = 16.4 Hz, 1H), 5.12 (dd, J = 9.5, 6.3 Hz, 1H), 6.22 (m, 1H), 6.97 (m, 2H), 7.06 (m, 2H), 7.11–7.14 (m, 2H), 7.20 (tm, J = 7.3 Hz, 2H), 7.24–7.31 (m, 3H), 7.40 (tm, J = 7.6 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) 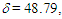 50.76, 52.26, 123.31, 125.03, 125.45, 126.57, 126.78, 128.27, 128.62, 128.73 (2C), 129.83, 129.96, 135.95, 144.44, 147.89, 201.15 ppm; UV (CH3OH)
50.76, 52.26, 123.31, 125.03, 125.45, 126.57, 126.78, 128.27, 128.62, 128.73 (2C), 129.83, 129.96, 135.95, 144.44, 147.89, 201.15 ppm; UV (CH3OH) 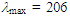
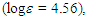 219sh (4.40), 233sh (4.13), 293 (3.78), 317sh (3.70), 361sh (2.99) nm; MS m/z (rel. int) 298 (M+, 12), 256 (12), 192 (11), 191 (13), 179 (18), 178 (28), 167 (16), 165 (21), 118 (100), 115 (12), 105 (57), 91 (14), 90 (81), 89 (14). Elemental analysis calcd for C22H18O C, 88.56, H 6.08%; found C, 88.78, H, 6.34%.
219sh (4.40), 233sh (4.13), 293 (3.78), 317sh (3.70), 361sh (2.99) nm; MS m/z (rel. int) 298 (M+, 12), 256 (12), 192 (11), 191 (13), 179 (18), 178 (28), 167 (16), 165 (21), 118 (100), 115 (12), 105 (57), 91 (14), 90 (81), 89 (14). Elemental analysis calcd for C22H18O C, 88.56, H 6.08%; found C, 88.78, H, 6.34%.3.7. 1,1-Diphenyl-1,4-Dihydroazulene (14) via TosylHydrazone of 12
- To a solution of 12 (900 mg, 3.02 mmol) in 45 mL of THF was added tosylhydrazine (560 mg, 3.00 mmol) was added. This mixture was stirred at room temperature under nitrogen for 4 days. Solids of tosylhydrazine gradually dissolved and the mixture became a clear solution after one day. Then after, the hydrazone product slowly crystallized and were collected by a suction filtration. After being washed with a trace amount of cold THF and well with ether to give 1.20 g (86% yield) of the tosylhydrazone of 12 as brownish microcrystals.Tosylhydrazone of 12: M.p. 173–176°C (dec.); IR (KBr)
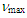 = 1421 (m), 1334 (m), 1185 (m), 1170 (s), 701 (s), 667 (s) cm–1; 1H NMR (CDCl3, 400 MHz)
= 1421 (m), 1334 (m), 1185 (m), 1170 (s), 701 (s), 667 (s) cm–1; 1H NMR (CDCl3, 400 MHz) 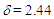 (s, 3H), 2.87 (d, J = 6.4 Hz, 2H), 3.21 (s, 2H), 5.61 (dt, J = 10.0, 6.4 Hz, 1H), 6.12 (dd, J = 10.0, 6.0 Hz, 1H), 6.21 (d, J = 11.5 Hz, 1H), 6.50 (dd, J = 11.5, 5.9 Hz, 1H), 7.02–7.05 (m, 4H), 7.18–7.28 (m, 6H), 7.32 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz)
(s, 3H), 2.87 (d, J = 6.4 Hz, 2H), 3.21 (s, 2H), 5.61 (dt, J = 10.0, 6.4 Hz, 1H), 6.12 (dd, J = 10.0, 6.0 Hz, 1H), 6.21 (d, J = 11.5 Hz, 1H), 6.50 (dd, J = 11.5, 5.9 Hz, 1H), 7.02–7.05 (m, 4H), 7.18–7.28 (m, 6H), 7.32 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) 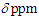 = 21.66, 23.03, 47.11, 59.23, 126.43, 126.65, 126.84, 127.87, 127.91, 128.03, 128.19, 128.22, 128.33, 128.45, 128.51, 129.57, 135.24, 144.20, 145.84 ppm; MS m/z (rel. int) 466 (M+, 7), 312 (25), 311 (100), 256 (6), 203 (6), 191 (8), 91 (12). HRMS calcd for C29H26N2O2S 466.1715; found, 466.1719.To a suspension of 466 mg (1.00 mmol) of the tosylhydrazone in 5 mL of THF was added a 1M methyllithium solution in ether (10 mL, 10 mmol) at 0°C under nitrogen atmosphere. The resulted orange solution was stirred at room temperature for 5 h and then was quenched by adding water. The mixture was extracted with ether (20 mL x 3) and the combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography (AcOEt/hexane = 2/98) to give 195 mg (69%) of 14 as a pale yellow oil.14: IR (liq. film)
= 21.66, 23.03, 47.11, 59.23, 126.43, 126.65, 126.84, 127.87, 127.91, 128.03, 128.19, 128.22, 128.33, 128.45, 128.51, 129.57, 135.24, 144.20, 145.84 ppm; MS m/z (rel. int) 466 (M+, 7), 312 (25), 311 (100), 256 (6), 203 (6), 191 (8), 91 (12). HRMS calcd for C29H26N2O2S 466.1715; found, 466.1719.To a suspension of 466 mg (1.00 mmol) of the tosylhydrazone in 5 mL of THF was added a 1M methyllithium solution in ether (10 mL, 10 mmol) at 0°C under nitrogen atmosphere. The resulted orange solution was stirred at room temperature for 5 h and then was quenched by adding water. The mixture was extracted with ether (20 mL x 3) and the combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography (AcOEt/hexane = 2/98) to give 195 mg (69%) of 14 as a pale yellow oil.14: IR (liq. film) 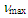 = 3059m, 3021m, 1597m, 1489m, 786s, 761s, 731s, 699s cm–1; 1H NMR (CDCl3, 400 MHz)
= 3059m, 3021m, 1597m, 1489m, 786s, 761s, 731s, 699s cm–1; 1H NMR (CDCl3, 400 MHz) 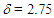 (d, J = 6.5 Hz, 2H), 5.43 (dt, J = 9.6, 6.5 Hz, 1H), 6.15 (dd, J = 9.8, 5.6 Hz, 1H), 6.26 (d, J = 5.3 Hz, 1H), 6.41 (dd, J = 11.2, 5.6 Hz, 1H), 6.48 (d, J = 11.2 Hz, 1H), 6.79 (d, J = 5.3 Hz, 1H), 7.09–7.12 (m, 4H), 7.17–7.25 (m, 6H) ppm; 13C NMR (CDCl3, 100 MHz)
(d, J = 6.5 Hz, 2H), 5.43 (dt, J = 9.6, 6.5 Hz, 1H), 6.15 (dd, J = 9.8, 5.6 Hz, 1H), 6.26 (d, J = 5.3 Hz, 1H), 6.41 (dd, J = 11.2, 5.6 Hz, 1H), 6.48 (d, J = 11.2 Hz, 1H), 6.79 (d, J = 5.3 Hz, 1H), 7.09–7.12 (m, 4H), 7.17–7.25 (m, 6H) ppm; 13C NMR (CDCl3, 100 MHz) 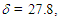 68.8, 121.1, 126.6, 127.6, 127.7, 128.0, 128.19, 128.23, 132.5, 136.1, 142.0, 147.0, 148.2 ppm; UV (CH3OH)
68.8, 121.1, 126.6, 127.6, 127.7, 128.0, 128.19, 128.23, 132.5, 136.1, 142.0, 147.0, 148.2 ppm; UV (CH3OH) 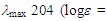 4.38), 243 (3.69), 328 (3.50) nm; MS m/z (rel. int) 282 (M+, 100), 281 (34), 268 (65), 267 (86), 266 (20), 265 (42), 252 (29), 205 (25), 204 (29), 203 (38), 202 (39), 191 (65), 189 (29). HRMS calcd for C22H18 282.1409, found, 282.1411.
4.38), 243 (3.69), 328 (3.50) nm; MS m/z (rel. int) 282 (M+, 100), 281 (34), 268 (65), 267 (86), 266 (20), 265 (42), 252 (29), 205 (25), 204 (29), 203 (38), 202 (39), 191 (65), 189 (29). HRMS calcd for C22H18 282.1409, found, 282.1411.3.8. 1,1-Diphenyl-1H-azulenium Perchlorate (9)
- To a solution of 190 mg (0.674 mmol) of 14 in 2 mL of acetonitrile was added 231 mg (0.674 mmol) of trityl perchlorate. After being stirred at room temperature for 1 h, the reaction solution was concentrated to ca. 0.5 mL under vacuum. To this mixture was added 4 mL of ether. The solids formed were collected by a suction filtration and were recrystallized from dichloromethane-ether to give 155 mg (82% yield) of 9 as greenish yellow microcrystals.9: M.p. 113–116°C; IR (KBr)
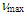 = 1508 (m), 1491 (m), 1450 (s), 1092 (br, s), 804 (m), 756 (m), 700 (m), 623 (s) cm–1; 1H NMR (CD3CN, 400 MHz)
= 1508 (m), 1491 (m), 1450 (s), 1092 (br, s), 804 (m), 756 (m), 700 (m), 623 (s) cm–1; 1H NMR (CD3CN, 400 MHz) 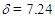 (dm, J = 7.1 Hz, 4H), 7.37 (tm, J = 7.1 Hz, 4H), 7.42 (tm, J = 7.1 Hz, 2H), 7.51 (d, J = 5.4 Hz, 1H), 8.01 (d, J = 5.4 Hz, 1H), 8.75 (m, 2H), 8.95 (tm, J = 9.6 Hz, 1H), 8.98 (dm, J = 9.6 Hz, 1H), 9.06 (d, J = 9.6 Hz, 1H) ppm; 13C NMR (CD3CN)
(dm, J = 7.1 Hz, 4H), 7.37 (tm, J = 7.1 Hz, 4H), 7.42 (tm, J = 7.1 Hz, 2H), 7.51 (d, J = 5.4 Hz, 1H), 8.01 (d, J = 5.4 Hz, 1H), 8.75 (m, 2H), 8.95 (tm, J = 9.6 Hz, 1H), 8.98 (dm, J = 9.6 Hz, 1H), 9.06 (d, J = 9.6 Hz, 1H) ppm; 13C NMR (CD3CN) 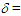 70.21, 128.94, 130.27, 130.47, 135.18, 137.55, 147.56, 149.31, 150.39, 150.75, 154.19, 164.15, 167.35, 172.54 ppm; UV-vis (CH3CN)
70.21, 128.94, 130.27, 130.47, 135.18, 137.55, 147.56, 149.31, 150.39, 150.75, 154.19, 164.15, 167.35, 172.54 ppm; UV-vis (CH3CN) 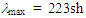
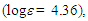 255 (4.30), 306 (3.88), 378 (3.76) nm; UV-vis (CH2Cl2)
255 (4.30), 306 (3.88), 378 (3.76) nm; UV-vis (CH2Cl2) 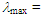 233sh
233sh 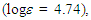 261 (4.67), 316 (3.98), 389 (3.76), 541sh (1.95) nm; MS m/z (rel. int) 281 (C22H17+, 34), 280 (100), 279 (18), 278 (19), 277 (14), 276 (14), 252 (12), 202 (13). HRMS calcd for C22H17+ 281.1325; found, 281.1327. Elemental analysis calcd for C22H17ClO4 C, 69.39, H 4.50%; found C, 69.33, H, 4.82%.
261 (4.67), 316 (3.98), 389 (3.76), 541sh (1.95) nm; MS m/z (rel. int) 281 (C22H17+, 34), 280 (100), 279 (18), 278 (19), 277 (14), 276 (14), 252 (12), 202 (13). HRMS calcd for C22H17+ 281.1325; found, 281.1327. Elemental analysis calcd for C22H17ClO4 C, 69.39, H 4.50%; found C, 69.33, H, 4.82%.3.9. Thermal Rearrangement of Cation 9
- An NMR tube was sealed with a solution of 9 (19.0 mg, 50.0 mmol) in 1.0 mL of CD3CN. The tube was spun and heated at 100°C inside the NMR spectrometer and the rearrangement reaction was monitored by the 1H NMR spectrum. After the reaction completed (210 min), the tube was cooled to room temperature and opened. The reaction mixture was poured into 20 mL of water and was extracted with chloroform (10 mL x 3). The combined organic layer was washed with a saturated NaHCO3 solution and brine. After dryness over MgSO4, the solvent was removed and the residue was purified by alumina chromatography to give 13.7 mg (98% yield) of 22 [20] as a greenish blue oil.1H NMR (CDCl3, 400 MHz)
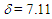 (t, J = 9.8 Hz, 1H), 7.16 (t, J = 9.8 Hz, 1H), 7.23–7.45 (m, 10H), 7.53 (t, J = 9.8 Hz, 1H), 7.58 (s, 1H), 8.25 (d, J = 9.8 Hz, 1H), 8.34 (d, J = 9.8 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz)
(t, J = 9.8 Hz, 1H), 7.16 (t, J = 9.8 Hz, 1H), 7.23–7.45 (m, 10H), 7.53 (t, J = 9.8 Hz, 1H), 7.58 (s, 1H), 8.25 (d, J = 9.8 Hz, 1H), 8.34 (d, J = 9.8 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz) 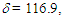 123.3, 123.6, 123.8, 126.5, 127.3, 128.2, 128.3, 128.4, 129.9, 131.4, 131.7, 135.5, 136.5, 136.7, 137.3, 139.5, 140.2 ppm; MS m/z (rel. int) 280 (M+, 100), 279 (22), 268 (44), 267 (54), 266 (26), 252 (20), 205(46), 203 (60), 202 (68), 192 (17), 189 (36), 165 (27), 126 (30), 91 (25), 77 (34), 51 (39). HRMS calcd for C22H16 280.1252; found, 280.1258.
123.3, 123.6, 123.8, 126.5, 127.3, 128.2, 128.3, 128.4, 129.9, 131.4, 131.7, 135.5, 136.5, 136.7, 137.3, 139.5, 140.2 ppm; MS m/z (rel. int) 280 (M+, 100), 279 (22), 268 (44), 267 (54), 266 (26), 252 (20), 205(46), 203 (60), 202 (68), 192 (17), 189 (36), 165 (27), 126 (30), 91 (25), 77 (34), 51 (39). HRMS calcd for C22H16 280.1252; found, 280.1258.3.10. Determination of pKR+ Value of Cation 9
- The UV spectra in various pH of 50% aqueous acetonitrile solutions were measured by exactly the same method of Komatsu et al. [23–24] Observed absorbance at the longest absorption maxima at 378 nm was plotted against pH to give a classical titration curve, whose midpoint was taken as the pKR+.
4. Conclusions
- We have accomplished the synthesis of the title compound, 1,1-diphenyl-1H-azulenium perchlorate (9), from 1-acetyl-1,3,5-cycloheptatriene (10). The compound 9 shows less stability than 8 compared with their pKR+ values and upon heating 9 undergoes rearrangement to produce 1,2-diphenylazulene. Further studies on synthesis of 1,1-diphenyl-1H-azuleniums having substituents on the phenyl rings are in progress to evaluate an effect of the substituents on stability of the cationic system.
ACKNOWLEDGEMENTS
- A financial support from the Faculty of Science in Shinshu University (for M.O.) is greatly acknowledged.