-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2015; 5(5): 149-152
doi:10.5923/j.ajoc.20150505.02

Solid-Liquid Phase Transfer Catalysis and Microwave-Assisted Green Synthesis of Tetracyclone
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLUrbain C. Kasséhin1, 2, Fernand A. Gbaguidi1, 3, Pascal Carato4, Christopher R. McCurdy5, Georges C. Accrombessi3, Jacques H. Poupaert2
1Medicinal Organic Chemistry Laboratory, School of Pharmacy, Faculté des Sciences de la Santé, Université d'Abomey-Calavi, Campus du Champ de Foire, Cotonou, Bénin
2Medicinal Chemistry Research Group (CMFA), Louvain Drug Research Institute, UCLouvain, Brussels, Belgium
3Laboratoire de Chimie Organique, Physique et de Synthèse, Faculté des Sciences et Techniques, Université d'Abomey-Calavi 01, Cotonou, Bénin
4Université de Poitiers, IC2MP - CNRS 7285, UFR de Médecine et de Pharmacie, 6 rue de la Milétrie, France
5Pharmacology and Medicinal Chemistry, Department of BioMolecular Sciences, School of Pharmacy, University of Mississipi, USA
Correspondence to: Urbain C. Kasséhin, Medicinal Organic Chemistry Laboratory, School of Pharmacy, Faculté des Sciences de la Santé, Université d'Abomey-Calavi, Campus du Champ de Foire, Cotonou, Bénin.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A green synthesis of tetracyclone (2, 3, 4, 5-tetraphenyl-2, 4-cyclopentadien-1-one) has been conceived and executed on the basis of a rationale design. Three separate, “green” processes to synthesize tetracyclone were undertaken to compare homogeneous and heterogeneous reaction conditions as well as to examine the efficacy of microwave enhancement and the utility of phase transfer catalysis. Our mechanistically-based approach involves a unique solid-liquid phase transfer catalysis, microwave activation, and nucleophilic catalysis involving anthranilic acid as a green catalyst. Use of these methods collectively resulted in a yield of 82% for the title compound with a minimal resultant waste stream.
Keywords: Tetracyclone (2, 3, 4, 5-tetraphenyl-2, 4-cyclopentadien-1-one), Microwave-Activation, Tetracyclone thiosemicarbazone derivatives, Diels–Alder-Reaction, Nucleophilic catalysis
Cite this paper: Urbain C. Kasséhin, Fernand A. Gbaguidi, Pascal Carato, Christopher R. McCurdy, Georges C. Accrombessi, Jacques H. Poupaert, Solid-Liquid Phase Transfer Catalysis and Microwave-Assisted Green Synthesis of Tetracyclone, American Journal of Organic Chemistry, Vol. 5 No. 5, 2015, pp. 149-152. doi: 10.5923/j.ajoc.20150505.02.
Article Outline
1. Introduction
- In the context of a medicinal chemistry research program centered on phenytoin analogs specifically designed for their neuroprotective properties potential, we became interested in the Biltz hydantoin synthesis [1-3]. This multi-component reaction allows creating a rather complex structure in a single operation and is amenable to the elaboration of a large compound library. Our group recently uncovered an imidazolone species as a pivotal intermediate, which shares structural similarities with tetracylone (i.e. 2, 3, 4, 5-tetraphenyl-2, 4-cyclopentadien-1-one, 1). This peculiar structure drew our attention because of its anti-aromatic character in its dienic mesomeric form (See structure 2 in Figure 1). Indeed, so far, the naked cyclopentadienone has remained a rather elusive structure due to its intrinsic instability. [4].
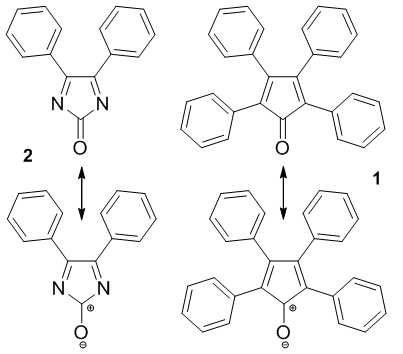 | Figure 1. Isoeletronic structures of tetracyclone (1) and imidazolone (2) derivative |
2. Results and Discussion
- Phase Transfer Catalysis (PTC) appears by far to be the most general, efficient and environment-friendly methodology of performing reactions involving the reaction of organic and inorganic anions with organic substrates. According to this methodology, the reactions are generally performed in immiscible two-phase systems. One phase (inorganic) is a source of inorganic anions (if they are available as salts) or base for generation of organic anions. The salts or bases are most often used as aqueous solutions, or less frequently in the form of finely divided solids. The second (organic) phase contains organic reagents, usually neat or sometimes dissolved in appropriate solvents. To obtain faster reaction rates, one should use the most concentrated reaction medium possible and therefore solid-liquid PTC conditions were explored. Solid-liquid phase transfer catalysis (PTC) without added solvent was shown to efficiently promote several condensation reactions but these had to be classically carried out generally in presence of toxic quaternary salts or crown-ethers phase-transfer catalysts. An utmost promising alternative is to replace these adjuvants by polyethylene glycol (PEG) oligomers.Indeed, since their first introduction, the popularity of the PT-catalysts has grown up enormously and, due to their inexpensive character, still nowadays PEG's are sometimes referrred to as "poor chemist's crown ether". However, the PEG structure is rather unique in its exceptional ability to behave as an anion-activating PT-catalyst, causing e.g. significant activation of hydroxide or acetate anions. But, not only are PEG's rather inexpensive, they are also endowed with very low human and environmental toxicity, which is of paramount importance in the framework of a green synthesis design.
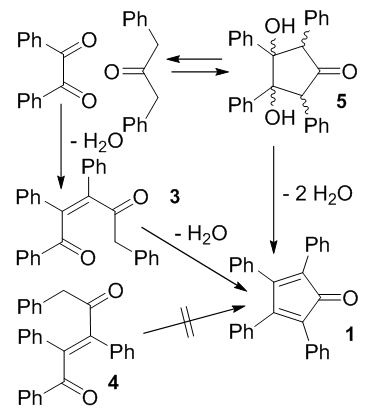 | Figure 2. Mechanism of formation of tetracyclone |
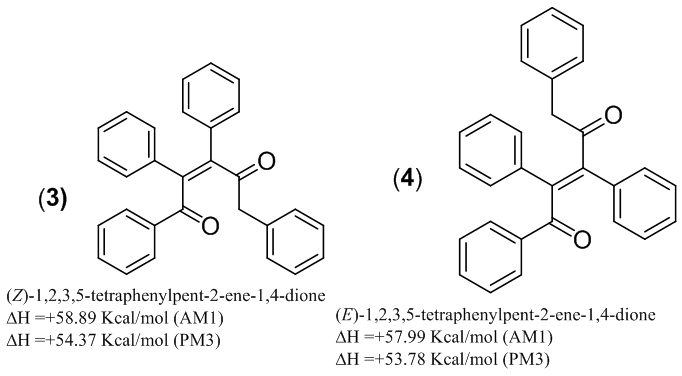 | Figure 3. Comparison thermodynamic of stability of 3 and 4 |
3. Experimental
3.1. Instrumentation
- Melting points were determined using an electrothermal melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer 457 spectrometer using KBr disks. Wave numbers are expressed in cm-1. 1H-NMR and 13C-NMR spectra were recorded at ambient temperature on a Bruker 400 spectrometer. Compounds were dissolved in CDCl3 or DMSO-d6. Chemical shifts are expressed in the δ scale with TMS (tetramethylsilane) as internal standard. Thin layer chromatography (TLC) analyses were performed on Merck TLC plates (silica gel, 60 F 254, E. Merck, Darmstadt, Germany, ref. 5735). For TLC, all the compounds reported here were routinely checked in two standard solvents, i. e. acetone/toluene/cyclohexane (solvent A, 5:2:3, v/v/v) and ethyl acetate/n-hexane (solvent B, 4:6, v/v). The reverse-phase thin layer chromatography conditions were: HPTLC plates RP-18 F-254 S (Merck), methanol: water (75/25, v/v). Details regarding semi-empirical quantum calculations are available from the corresponding authors.All compounds reported were found homogenous under such TLC and HPTLC conditions. All reagents were purchased from Aldrich (Milwaukee, USA). All solvents were of the ACS reagent grade (Aldrich). Initial exploratory small-scale microwave experiments were conducted using a CEM Discover Synthesis Unit (monomode system) operating at 2450 MHz and monitored by a PC computer. Microwaves produced in this device are focussed by a circular wave-guide all around the cavity. The temperature was measured by optical fibre or infrared detection with continuous-feedback temperature control, and maintained at a constant value by power modulation (0−300 W). Stirring was provided by an in situ magnetic variable speed stirrer when reactions were performed in closed vessels under controlled pressure or with mechanical stirring in the case of reactions in open vessels. Reactions were performed either in glass vessels (capacity 10 mL) sealed with a septum or in open vessels (capacity 100 mL). The pressure was controlled by a load cell connected to the vessel via a needle, which penetrated just below the septum surface.
3.2. General Conditions
3.2.1. Synthesis of 2, 3, 4, 5-Tetraphenyl-2, 4-Cyclopentadien-1-One (Homogenous Conditions)
- In a 500-mL round-bottomed flask, 21 g (0.1 mol) of benzil and 21 g. (0.1 mol) of dibenzyl ketone are dissolved in 250 mL of hot ethanol. The flask is fitted with a reflux condenser, the temperature of the solution is raised nearly to the boiling point, and a solution of 3 g of potassium hydroxide in 15 mL of ethanol is added cautiously in several portions through the reflux condenser. When the effervescence has subsided, the mixture is refluxed for another 30 minutes and then cooled to 0° in an ice bath. The dark crystalline product is filtered with suction and washed with three 10-ml. portions of 95% ethanol. The product melts at 218–219°C and weighs 35 g (Yield = 91%). The rough product can be crystallized from a mixture of absolute ethanol and toluene and the melting point of the final pure material (TLC, NMR) is 219–220°C with important loss of material (Final yield = 62%). Spectral data were in all respects identical to a commercial sample of the product (Acros Organics).
3.2.2. Synthesis of 2, 3, 4, 5-Tetraphenyl-2, 4-Cyclopentadien-1-One (1, Homogenous Conditions under Microwave Activation)
- The microwave oven used for this large sale preparation was a commercial household microwave oven (Moulinex® FM1935G, frequency: 2450 MHz). In a 250-mL erlenmeyer flask, 25 mL of 1.2 M aqueous KOH were slowly added at room temperature to a mixture of 20.2 g of benzil (96.2 mmol) and 21.0 g of dibenzylketone (100 mmol) dissolved in 40 ml of DMSO. Some heat evolution was initially noted. Following an initial 90 sec 750 W pulse, the mixture was stirred for 5 min; 30 sec pulses were then applied at 6, 9, 12, 15, 18, 21, 24, and 30 min, the mixture was stirred between pulses. The mixture was then poured under vigourous stirring into 300 mL of ice-cold water. The precipitate was filtered over a Buchner funnel. The dark colored precipitate so collected was dried in vacuo overnight and recrystallized from 95% ethanol. Yield = 74%. Mp 219–220°C. Spectral data were identical to a commercial sample of the product (Acros Organics)
3.2.3. Synthesis of 2, 3, 4, 5-Tetraphenyl-2, 4-Cyclopentadien-1-One (1, Heterogenous Conditions under Microwave Activation)
- A slurry of 5 mmol of benzil and 5 mmol of anthranilic acid in 1 mL of PEG400 was irradiated and stirred for 1 min at 150 W power using a CEM Discover Synthesis Unit (See 3.1 Instrumentations). To this mixture were added 5 mmol of dibenzylketone had subsided, the reaction mixture was once again irradiated and stirred for 1 min at 150 W power. The dark colored system and 10 mmol of KOH in pellets. After the frothing was allowed to cool down to room temperature and treated with 20 mL of distilled water. Workup and purification were performed as above. After recrystallization from ethanol, the yield was 82%. Melting point, TLC, and spectral data were in all respects identical to a commercial sample of the product (Acros Organics).
4. Conclusions
- In this paper, the authors revisited the synthesis of tetracyclone. They noted that this process is best carried out using solid-phase transfer catalysis and microwave activation. Further work is now performed along this line to generate dendromeric hexaphenylbenzene species derived from tetracyclone as well as thiosemicarbazone derivatives synthesized with a view to their potential trypanocidal activity. This work emphasizes also the beneficial role of nucleophilic catalysis in the context of the generalized acid-base catalysis [10]. Put together, all these technologies provide a green procedure for an expedited high-yield synthesis of tetracyclone and related derivatives allowing to obtain a final yield of 82%.
ACKNOWLEDGEMENTS
- The authors wish to acknowledge the generous contribution of the CTB/BTC (Coopération Technique Belge, Brussels, Belgium) both in terms of fellowship (U.C.K.) and financial support.