-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2015; 5(5): 137-148
doi:10.5923/j.ajoc.20150505.01

Synthesis, Biological and Anti-Tumor Evaluation of Some New Nucleosides Incorporating Heterocyclic Moieties
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFekria M. A. Soliman, Nadia T. A. Dawoud, Rehab M. Hamza
Department of Chemistry, Faculty of Science (Girls), Al-Azhar University, Cairo, Egypt
Correspondence to: Nadia T. A. Dawoud, Department of Chemistry, Faculty of Science (Girls), Al-Azhar University, Cairo, Egypt.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

1,3-diaryl-1-propen-3-ones 1a-h, were used as building blocks for a large range of nucleoside analogs incorporating five and six-membered heterocyclic rings. Heterocyclic compounds incorporating aromatic moieties (2-11) and their N-nucleoside analogs (13-20) were synthesized. New compounds were evaluated for their potential antimicrobial and antifungal activities and for their in vitro cytotoxic activity against three cell lines: human breast cancer cell line (MCF-7), colon carcinoma cells (HCT) and human epidermid/arynx carcinoma cell line (HEp2).
Keywords: Chalcones, Heterocyclic compounds, N-Nucleosides, Antimicrobial and anticancer activities
Cite this paper: Fekria M. A. Soliman, Nadia T. A. Dawoud, Rehab M. Hamza, Synthesis, Biological and Anti-Tumor Evaluation of Some New Nucleosides Incorporating Heterocyclic Moieties, American Journal of Organic Chemistry, Vol. 5 No. 5, 2015, pp. 137-148. doi: 10.5923/j.ajoc.20150505.01.
Article Outline
1. Introduction
- Heterocyclic compounds occur widely in nature. Nitrogen-containing heterocyclic molecules constitute the largest portion of these chemical entities, which are part of many natural products. Indazole derivatives are interesting compounds, with many having biological as well as pharmaceutical activity [1-3]. Some new indazole derivatives were investigated as electronically active materials [4-9]. Cyanopyridone and cyanopyridine derivatives have promising antimicrobial activities [10, 11] as well as anti-cancer activities [12-14]. Oxazine derivatives represent an important classes of organic compounds; 1,3-oxazines in particular have been extensively studied because of their profound biological activities including antibacterial [15, 16], antifungal [17], antituberculor [18], antitumor and anti-HIV agents[19, 20]. 1,3-Oxazine derivatives are also known as progesterone receptor agonists [21]. Pyrimidine derivatives are very well known in medicinal chemistry for their therapeutic applications [22, 23]. One important class of pyrimidine is 2-thiopyrimidine and its derivatives, which are also well known as 2-mercaptopyrimidine compounds [24, 25]. Carbohydrates are ubiquitous in nature, readily available, cheap, biodegradable and non-toxic materials [26, 27]. Presence of several functional groups and stereogenic centers in carbohydrates permit stereochemical differentiations, enantiopure compound synthesis [28-30], use as chiral templates [31], biosensors [32] and as precursors for several biologically active products [33]. Besides these crucial roles, carbohydrates possess a unique set of chemical and structural feature that make them particularly attractive as molecular scaffolds. The aim of this work was to design, synthesis of indazole, pyridines, 2-aminooxazines and/or 2-thiopyrimidine derivatives bearing aromatic moieties and use these compounds as a basis for the synthesis of a series of nucleosides. Some of the new compounds were then examined for cytotoxic activity via assays on human breast cancer cell line MCF-7, colon carcinoma cells (HCT), human epidermid/arynx carcinoma cell line (HEp2).
2. Results and Discussion
- 1,3-diaryl-1-propen-3-ones, 1a-h, which were synthesized according to the literature [34], were used as starting material for the synthesis of a large range of heterocyclic compounds (compound series 2-6) as depicted in Scheme 1.
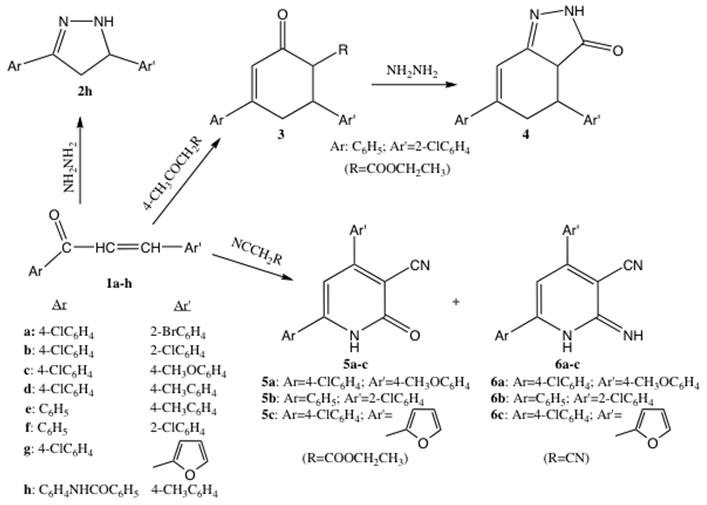 | Scheme 1. Heterocycle synthesis from 1,3-diaryl-1-propen-3-ones |
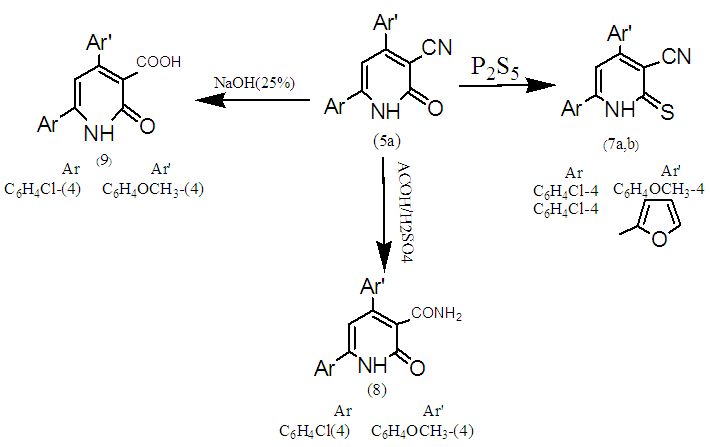 | Scheme 2. Functionalizations of 5a,c |
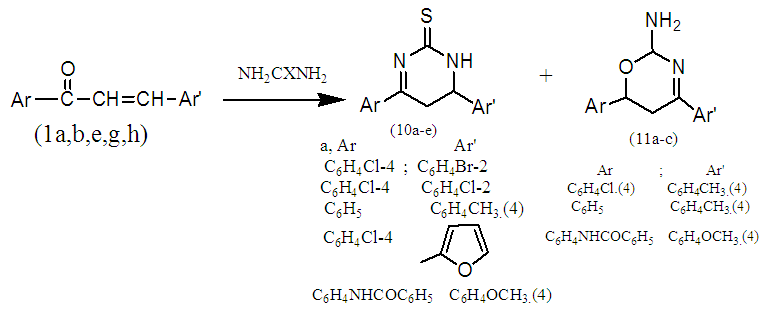 | Scheme 3. Heterocyclization reactions with 1a,b,e,g,h |
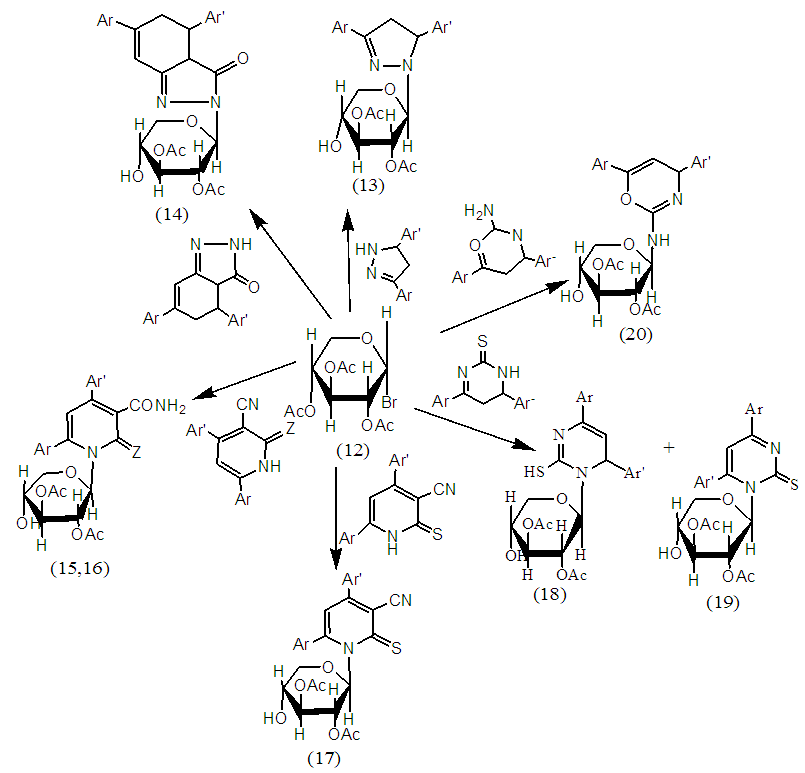 | Scheme 4. Reaction of Compound 12 with Heterocycles |
3. Biological Evaluation
3.1. Antimicrobial Activity
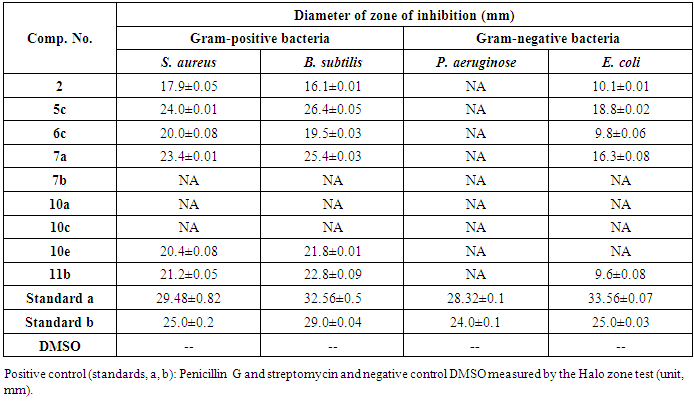
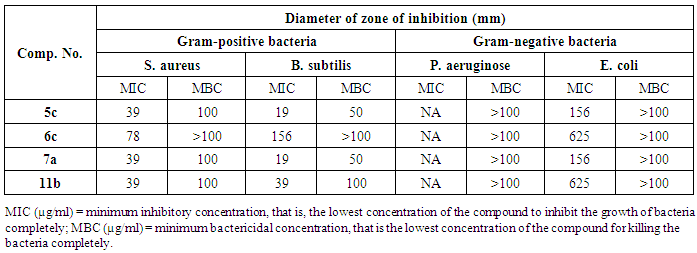
- Previously untested compounds were evaluated for antimicrobial activity against eight strains of microorganisms using the agar diffusion technique. The tested compounds were screened against two Gram-positive bacteria, Staphylococcus aureus (RCMB 000106) Bacillus subtilis (RCMB 000107); two Gram-negative bacteria, Pseudomonas aeruginosa (RCMB 000102), Escherichia coli (RCMB 000103) and four fungi, Aspergillus fumigatus (RCMB 002003), Geotrichum candidum (RCMB 052006), Candida albicans (RCMB 005002) and Syncephalastrum racemosum (RCMB 005003) by the disk diffusion method. Penicillin G. Streptomycin were used as positive control for bacterial strains while, Itraconazole and Clotrimazole were used as positive controls for the fungi strains. The investigation of antibacterial screening data revealed that compounds 5c and 10e were the most potent towards the Gram-positive bacteria S. aureus and B. subtilis. Compounds 6c, 10e and 11b showed good to moderate activity against Gram-positive bacteria S. aureus and B. subtilis. Compound 2 was less potent, while 7b, 10a were inactive. As for the bacterial inhibition of the Gram-negative bacteria, the screening data showed that compounds, 5c and 7a were the most potent against E. coli. Compounds 2, 6c and 11b showed a relatively poor inhibition towards E. coli. All the tested analogs showed no activity against P. aeruginosa. Similarly, compounds 7b and 10a,c,e were inactive against the Gram-negative bacteria P. aeruginosa or E. coli. The bacterial zone of inhibition values are given in Table 1.
|
|
3.2. Antifungal Studies
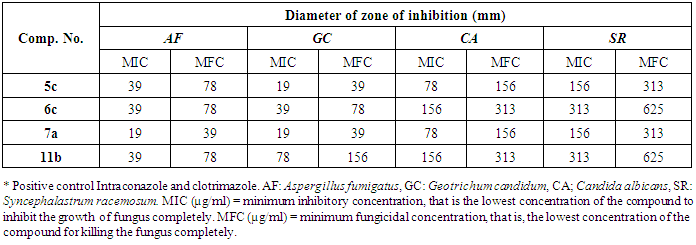
- Antifungal activity testing was also done by the disk diffusion method [43]. For assaying antifungal activity Aspergillus fumigatus (RCMB 002003), Geotrichum candidum (RCMB 052006), Candida albicans (RCMB 005002) and Syncephalastrum racemosum (RCMB 005003) were recultured in DMSO by the agar diffusion method. The lowest concentration (highest dilution) required to arrest the growth of fungus was regarded as minimum inhibitory concentration (MIC). The minimum inhibitory concentration and minimum fungicidal concentration are given in Table 3.
|
3.3. Cytotoxicity Studies
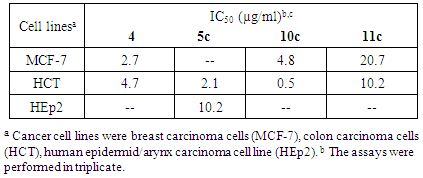
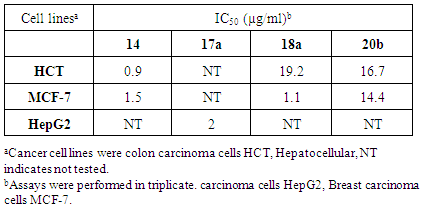
- Cytotoxicity tests were performed using compounds 4, 5c, 10c and 11c against three cancer cell lines, breast cancer cell line MCF-7, colon carcinoma cells (HCT), human epidermid/arynx carcinoma cell line (HEp2) by using a modified method [35]. The results (Table 4) showed that 4 had slight activity toward the HCT cell line (IC50 = 4.7 µg/ml) and its activity towards MCF-7 cell line was lower (IC50 = 2.7 µg/mL). Compound 11c exhibited cytotoxic activity against the HCT cell line (IC50 10.2 µg/ml) and a higher cytotoxic activity against MCF-7 with IC50 = 20.7 µg/ml. The cytotoxic activity of 5c towards HEP-2 was moderately potent with an IC50 = 10.2 µg/ml while its cytotoxic activity against colon carcinoma cells was very low with an IC50 = 2.1µg/ml. The cytotoxic activity of 10c towards the MCF-7 cell line was relatively weak with an IC50 = 4.8 µg/ml, while the cytotoxicity against HCT cell line was nearly inactive given the observed IC50 of 0.5 µg/ml.
|
|
4. Conclusions
- In summary, we have synthesized a novel series of nucleoside analogs in moderate to high yields. The prepared compounds which contain the pyridine-3-carbonitrile moiety provide better antimicrobial activity against S. aureus, B. subtilis than similar molecules without this functionality. Most of the prepared compounds revealed potential anticancer activity against the colon cancer cell line, Hepatocellular cancer cell line, Breast cell line and epidermid / arynx cancer cell line. Compounds 4, 5c, 10c, 11c, 14, 17a, 18a and 20b exhibited good antitumor activity when compared with the reference drug.
5. Experimental
- All melting points for the prepared derivatives were measured in capillary tubes using a Gallen-Kamp apparatus and were uncorrected. The IR spectra were recorded on a Perkin-Elmer 1650 spectrophotometer (KBr pellets) and the wave numbers were given in cm-1. The 1H, 13C NMR spectra were measured in dimethyl sulphoxide-d6 as a solvent using a Varian Gemini 180 spectrometer operating at 300 MHz for 1H, and 75 MHz for 13C. TMS was used as an internal standard and the chemical shifts were reported as δ ppm. The FAB mass spectra were recorded on a JEOL SX 102/DA-6000 mass spectrometer. Synthesis of N-(4-(5-(4-methoxyphenyl)-4,5-dihydro-1H- pyrazol-3-yl)phenyl)benzamide (2).A mixture of (E)-N-(4-(3-(4`-methoxyphenyl) acryloyl) phenyl) benzamide (1h) (0.01mol) and hydrazine hydrate (0.01mol) in 30 ml of ethanol was refluxed for 6h. The yellow precipitate formed after cooling was filtered off, dried and recrystallized from ethanol to afford the required product (2) as yellow crystals, in 75% yield, m.p. 181C; [Requires: C, 74.39; H, 5.66; N, 11.32; Found: C, 74.35; H, 5.7; N, 11.4. IR (cm-1): 3340, 2942, 2846, 1650, 1600, 1510.Synthesis of ethyl 6-(4-chlorophenyl)-2-oxo-4- phenylcyclohex-3-ene carboxylate (3).A mixture of (1f) (0.01mol) and ethylacetoacetate (0.01mol) in 30ml of absolute ethanol containing sodium ethoxide (prepared from 0.2 g of sodium metal and 4.6 ml of absolute ethanol) was refluxed for 6h. After concentration and cooling the residue was poured into water, filtered off, washed well with dilute alcohol and recrystallized from ethanol to afford 3 as white crystals, in 60% yield, m.p 124°C. Requires: C, 71.08; H, 5.35; Cl, 10.01: Found: C, 71.1; H, 5.4; Cl, 10.1. IR (cm-1): 3006, 2928, 2818, 1698, 1660. MS (m/z, %); 355 (9.2%), 320 (3.8%), 308 (6.1%), 278 (34.6%), 249(1.6%), 192 (4.8%), 144 (100%).Synthesis of 4-(4-chlorophenyl)-6-phenyl-3,3a,4,5- tetrahydro-2H-indazolone (4).A mixture of 3 (0.01 mol) and hydrazine hydrate (0.01 mol) in 15 ml of acetic acid was heated under reflux for 6h. The solvent was evaporated and the product was collected, washed well with dilute ethanol and recrystallized from ethanol to give (4) as brown crystals, in 60% yield, m.p 144°C. Requires: C, 70.69; H, 4.65; N, 8.68; Cl, 11.007: Found: C, 71.0; H, 4.7; N, 8.7; Cl, 11.1IR (cm-1): 3392, 3216; 2918, 2846, 1670, 1604, 1508. MS (m/z, %); M+, M+2, M+3, 322, 324, 325 (99.4, 30.7, 4.6%).Synthesis of 4,6-diaryl-2-oxo-1,2-dihydropyridine-3- carbonitrile (5a-c). A mixture of 1c, 1f, and 1g (0.01 mol), ethylcyanoacetate (0.01 mol) and ammonium acetate (0.04 mol) in 30 ml of absolute ethanol was heated under reflux for 6h. It was then allowed to cool, filtered off, washed well with water, then with dilute alcohol, and recrystallized from ethanol to give the corresponding derivatives (5a-c) respectively.5a as yellow crystals, in 70% yield, m.p. 51°C. Requires: C, 67.75; H, 3.86; N, 8.32; Cl, 10.54: Found C, 67.6; H, 3.9; N, 8.4; Cl, 10.2; MS (m/z, %); 336.3 (96.0%) M+, 50 (100%). 1H-NMR spectrum (DMSO-d6): δ 3.83 (s, 3H, OCH3), 7.12-8.10 (m, 8H,C=CH and Ar-H) 8.28 (s, 1H, NH).5b as white crystals, in 70% yield, m.p. 80°C. Requires: C, 70.47; H, 3.58; N, 9.13; Cl, 11.58: Found C, 70.5; H, 3.6; N, 9.2; Cl, 11.6; IR (cm-1): 3460-3230, 2230-2210, 1654-1662.5c as brown crystals, in 75% yield, m.p. 60°C. Requires: C, 64.75; H, 3.03; N, 9.44; Cl, 11.97: Found: C, 64.5; H, 3.00; N, 9.5; Cl, 11.5; IR (cm-1): 3460-3230, 2230-2210, 1654-1662.Synthesis of 2-imino-4,6-diaryl-1,2-dihydropyridin-3- carbonitrile (6a-c).A mixture of 1c, 1f, and 1g (0.01 mol), malononitrile (0.01 mol) and ammonium acetate (0.04 mol) was fused on a sand-bath at 135-165°C for 3h. The product formed after cooling was washed with water, then with dilute ethanol, and recrystallized from the proper solvent to give (6a-c) respectively.6a as white crystals, in 71% yield, m.p. 52°C. Requires: C, 67.95; H, 4.17; N, 12.51; Cl, 10.58: Found: C, 68.0; H, 4.2; N, 9.5; Cl, 12.5; Cl, 10.6; IR (cm-1): 3100, 2220, 1662, 690 (C–Cl).6b as white crystals, in 70% yield, m.p. 75°C. Requires: C, 70.70; 3.92; N, 13.74; Cl, 11.62: Found: C, 70.7; H, 3.9; N, 13.8; Cl, 11.6; IR (cm-1): 3366, 2212, 1626, 686 (C–Cl).6c as black crystals in 60% yield, m.p. 70°C. Requires: C, 64.67; 3.38; N, 14.21; Cl, 12.01: Found: C, 65.0; H, 3.4; N, 4.1; Cl, 12.1; IR (cm-1): 3114, 2208, 1648, 640 (C–Cl).Synthesis of 4,6-diaryl-2-thioxo-1,2-dihydropyridin-3- carbonitrile derivatives (7a,b). A mixture of 5a and/or 5c (0.01mole) and P2S5 (0.01mol) in 15ml of dry xylene was refluxed for 6h. After cooling the solvent was evaporated under reduced pressure and the product was treated with petroleum ether (b.p. 40-60°C), then recrystallized from the proper solvent as 7a,b.7a as reddish brown crystals, in 70% yield, m.p. 200°C. Requires: C, 64.68; H, 3.68; N, 7.94; S, 9.07; Cl, 10.07: Found: C, 64.7; H, 3.7; N, 7.9; S, 9.1; Cl, 10.1; IR (cm-1): 3354, 3437, 1590, 1588, 1218, 1212. MS m/z, (%): 352.5 (9.82%), 324.2 (9.57%), 270 (10.18%), 241.2 (9.1%, 207.2 (10.55%), 171.2 (9.38%), 63 (100%).7b as dark brown crystals, in 60% yield, m.p. 110-112°C. Requires: C, 61.44; H, 2.88; N, 8.96; S, 10.24; Cl, 11.36: Found: C, 61.5; H, 2.9; N, 8.8; S, 10.2; Cl, 11.4; IR (cm-1): 3354, 3437, 1590, 1588, 1218, 1212.Synthesis of 6-(4-chlorophenyl)-4-(4-methoxyph-enyl)-2-oxo-1,2-dihydropyridin-3-carboxamide (8)A mixture of (5a) (1g) of and (5ml) of 30-40% H2SO4 in 15ml acetic acid was refluxed for 6hrs. The Precipitate was filtered off and recrystallized from benzene to give (8) as white crystals in 60% yield, m.p 120°C. Requires: C, 67.35; H, 4.43; N, 8.27; Cl, 10.48: Found: C, 67.1; H, 4.1; N, 8.1; Cl, 10.23; IR (cm-1): 3410, 3464, 1648.Synthesis of 6-(4-chlorophenyl)-4-(4-methoxyph-enyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (9).A mixture of (5a) (0.2 mol), ethyl alcohol (13 ml) and (3 ml) of 25% NaOH was stirred on magnetic stirrer, (10 ml) of 30% H2O2 was added gradually, the solution left to cool in ice bath. After an hour, the reaction was permitted to run at 50°C for an additional 3h. Then 5% sulphuric acid was added to neutralize the solution, the solvent was evaporated and solid product was recrystallized from benzene to give 9 as white crystals in 60% yield, m.p. 170°C. Requires: 67.16; H, 4.12; N, 4.12; Cl, 10.45: Found: C, 67.1; H, 4.1; N, 4.1; Cl, 10.23; IR (cm-1): 3433, 3200, 1686, 1590.Synthesis 4,6-diaryl-5,6-dihydropyrimidin-2(1H)-thione derivatives (10a-e).A mixture of (1a,b,f, g, and h) (0.01 mol) and thiourea (0.01mol) in 30ml ethanol containing (0.01mol) sodium ethoxide was heated under reflux for 6h. After concentration and cooling, the residue was diluted with water, filtered off, then washed well with warm water and dilute alcohol and recrystallised from the proper solvent to give (10a-e). 10a as pale white crystals in 70% yield, m.p. 200°C. Requires: C, 50.59; H, 3.16; N, 7.37; S, 8.43; Cl, 9.35; Br, 21.08: Found: C, 50.6; H, 3.2; N, 7.4; S, 8.4; Cl, 9.4; Br, 21.1; IR (cm-1): 3408, 3156, 2620, 1662, 1014, 454, 580 (C-Br, C-Cl).10b as brown crystals in 70% yield, m.p. 190°C. Requires: C, 57.31; H, 3.58; N, 8.35; S, 9.55; Cl, 21.19; Found: C, 57.3; H, 3.6; N, 8.4; S, 9.6; Cl,21.2; IR (cm-1): 3398, 3152, 2620, 1656, 1018, 580 (C-Cl).10c as yellow crystals in 70% yield., m.p. 135°C. Requires: C, 55.65; H, 3.76; N, 8.11; S, 9.27; Br, 23.18; Found: C, 55.7; H, 3.8; N, 8.2; S, 9.3; Br, 23.3; IR (cm-1): 3325, 3150, 2620, 1650, 1018, 580 (C-Cl).10d as dark brown crystals in 60% yield., m.p. 215°C. Requires: C, 57.83; H, 3.78; N, 8.63; S, 11.01; Cl, 12.22; Found: C, 57.9; H, 3.9; N, 9.7; S, 11.1; Cl, 12.3; IR (cm-1): 3210, 3140, 2706, 1650, 1012, 620 (C-Cl).10e as yellow crystals in 60% yield, m.p. 131°C. Requires: C, 69.39; H,5.06; N,10.1;S,7.71; Found: C, 69.4; H, 5.1; N, 10.2; S, 7.8; IR (cm-1): 3258, 1650, 1026.Synthesis of 2-amino-4,6-diaryl-2H-1,3-oxazine derivatives (11a-c).A mixture of 1d,f,h, (0.01 mol) and urea (0.01 mol) in 15 ml of absolute ethanol containing 6 ml of glacial acetic acid was heated under reflux for 6h. The precipitate that formed after cooling was collected, washed well with dilute ethanol and recrystallized from the proper solvent to give 11a-c, respectively.11a as Brown crystals in 60% yield, m.p. 194°C. Requires: C, 67.01; H, 5.23; N, 9.77;Cl, 12.39; Found C, 67.1; H, 5.3; N, 9.8;Cl, 12.4; IR (cm-1): 3348, 1652, 1210, 516.11b as white crystals in 70% yield, m.p. 109°C. Requires: C, 67.48; H, 4.56; N, 9.84; Cl, 12.47; Found: C, 67.5; H, 4.6; N, 9.9; Cl, 12.5; IR (cm-1): 3200, 1654, 1212, 686. 1H–NMR (DMSO-d6): δ 2.1 (s, 2H, NH2), 3.81, 3.82 (d, 1H, CHa), 6.9, 7.02 (d, 1H, CHb), 7.5-8.1 (m, 9H, Ar-H). 13C-NMR: δ 37.2 (C1), 161.1 (C2), 97.07 (C3), 168.8 (C4), 130.5 (C5), 127.7 (C6), 139.3 (C7), 143.4 (C8), 134.5 (C9), 127.3 (C10), 143.1 (C1`), 129.6 (C3`), 131.8 (C6`), 128.3 (C5`), 127.7 (C2`), 119.4 (C4`).11c as white crystals in 60% yield, m.p. 210°C. Requires: C, 72.18; H, 5.26; N, 10.52; Found: C, 72.2; H, 5.3; N, 10.6; IR (cm-1): 3286, 1662, 1260.Synthesis of nucleoside derivatives (13).A suspension of 2 (0.01 mol) in 6 ml of aqueous potassium hydroxide (prepared by dissolving 0.01 mol in 6 ml of distilled water) was stirred by using a magnetic stirrer for 3h, then a solution of 12 (0.0 mol) dissolved in 30 ml of dry acetone was added drop-wise while stirring which continued for 12 h. After evaporation of the solvent (reduced pressure), the residue was washed with dilute ethanol several times and the precipitate formed was recrystallised from ethanol to give 13 as brown crystals in 60% yield, m.p. 168°C. Requires: C, 64.92; H, 5.41; N, 7.32; Found: C, 65.0; H, 5.4; N, 7.2. IR (cm-1): 3346.2, 3000.9, 2840.3, 1663.6, 1600.1. 1H–NMR (DMSO-d6): δ 6.9, 7.0 (d, H-1' and H-2'), 2.41, 2.43 (s, 3H, 2xCOCH3), 10.04 - 10.52 (s, 1H, 2xOH). Synthesis of (2R,3S,4R,5S)-5-hydroxy-2-[3-oxo-(4-phenyl-5-(2-chloro-phenyl)-3,3a,4,5-tetrahydro-2H-indazol-2-yl)tetrahydro-2H-pyran-3,4-diyl diacetate (14).A suspension of 4 (0.01 mol) in 6ml of aqueous potassium hydroxide (prepared by dissolving 0.01 mol in 6ml of distilled water) was stirred using a magnetic stirrer for 3 h, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring, which continued for 12 h. After evaporation of the solvent (reduced pressure), the residue left was washed with dilute ethanol (several times) and the precipitate formed was re-crystallized from ethanol to give 14 as grey crystals, in 60% yield, m.p.102°C. Requires: C, 62.51; H, 4.83; N, 5.21; Cl, 6.60; Found: C, 62.5; H, 4.8; N, 5.3; Cl, 6.7; IR (cm-1): 3443, 1742, 1606 and 1376.Synthesis of nucleoside derivatives 15 and 16.A suspension of the cyanopyridone derivatives 5c and/or 2-imino-cyanopyridine 6c (0.01 mol) in 6ml of aqueous KOH solution (prepared from dissolving (0.01 mol) solid KOH in 6ml of distilled water) was well stirred (magnetic stirrer) at room temperature for 3 h, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring. Stirring was continued for further 12 h. After evaporation of the excess solvent (reduced pressure), the residue left was washed with dilute alcohol (several times) and the precipitate formed was recrystallized from ethanol to give 15 and 16.15 as brown crystals, in 70% yield, m.p. 70°C. Requires: C, 56.55; H, 4.34; N, 5.28; Cl, 6.69; Found: C, 56.6; H, 4.5; N, 5.4; Cl, 7.1; IR (cm-1): 3315.4, 1648, 1595, 525.8 (C-Cl). 1H–NMR (DMSO-d6): δ 4.41 (s, 1H, NH), 6.68-7.90 (m, 7H, Ar-H), 8.05 (d, 2H, 2xCH), 8.07(s, 1H, OH), 3.3, 3.41, 3.44 (t, 2H, CH2), 8.07 (s, 1H, NH2). MS (m/z, %): 497.45 (1.1%) M+, 461 (1.05%), 232 (15.4%), 190 (1.09%), 148 (1.37%).16 as black crystals in 60 % yield, m.p. 76°C. Requires: C, 56.65; H, 4.53; N, 7.93; Cl, 6.7; Found: C, 56.87; H, 4.67; N, 8.1; Cl, 6.9; IR (cm-1): 3314 (br), 1655, 1584, 526 (C-Cl). 1H-NMR (DMSO-d6): δ 1.97, 2.49 (s, 3H, 2xCOCH3), 6.55 (s, 1H, NH), 2.44, 3.44 (d, 2H, 2xCH), 6.82-7.94 (m, 7H, Ar-H), 8.12 (s, 2H, NH2) , 9.75 (s, 1H, OH). MS (m/z, 511.5 (0.4%), 484.5 (0.4%), 473.5 (0.69%), 426 (0.45%) and the base peak at m/z 50.Synthesis of 2R, 3S, 4R, 5S-2-(4,6-diaryl-3-carbonitrile)-2-thioxo-1(2H)-pyridin-yl)-5'-hydroxy-tetrahydro-2H-pyran-3,4-di-yl) diacetate (17a,b). A suspension of the thiocyanopyridine derivatives 7a and/or 7b (0.01 mol) in 6ml of aqueous KOH solution (prepared from dissolving (0.01 mol) solid KOH in 6ml of distilled water) was well stirred (magnetic stirrer) at room temperature for 3 hrs, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring. Stirring was continued for further 12 hrs. After evaporation of the excess solvent (reduced pressure), the residue left was washed with dilute alcohol (several times) and the precipitate formed was recrystallized from ethanol as 17a,b.17a as red crystals, in yield 70% ,m.p 146°C.[Requires: C, 59.10; H, 4.39; N, 4.92; Cl, 6.24;S,5.62 ; Found C, 59.32; H, 4.5; N,5.10 ; Cl, 6.34;S,5.78 %]; IR cm-13436.5 ,2220.4, 1600.6,1248.6 and 526.4 for OH, C≡N,C=N,C=S and C-Cl.17b as black crystals, in 60% yield, m.p. 184°C. Requires: C, 56.76; H, 3.97; N, 5.29; Cl, 6.72; S, 6.05; Found: C, 56.87; H, 4.01; N, 5.43; Cl, 6.9; S, 6.32; IR (cm-1): 3300, 2228, 1597, 1232 and 525 (C-Cl).Synthesis of 2S,3S,4R,5S-2-(6-(4-chlorophenyl)-4-(2-bromophenyl)-2,3,4,5-tetrahydro-pyrimidin-1-yl)mercapto-5-hydroxy tetrahydro-2H-pyran-3,4-diyl diacetate (18a) and 2S,3S,4R,5S-2-(6-(phenyl)-4-(2-bromophenyl)-2,3,4,5-tetra-hydro-pyrimidin-1-yl)-mercapto-5-hydroxy tetrahydro-2H-pyran-3,4-diyl diacetate (18b). A suspension of the pyrimidin-2-thione derivatives (10a) and/or (10c) (0.01 mol) in 6ml of aqueous KOH solution (prepared from dissolving (0.01 mol) solid KOH in 6ml of distilled water) was well stirred (magnetic stirrer) at room temperature for 3 h, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring. Stirring was continued for further 12 h. After evaporation of the excess solvent (reduced pressure), the residue left was washed with dilute alcohol (several times) and the precipitate formed was recrystallized from ethanol to give (18a,b).18a as grey crystals , in 70% yield, m.p. 188°C. Requires: C, 50.46; H, 3.86; N, 4.71; S; 5.38; Cl, 6.24; Br, 13.4; Found: C, 50.52; H, 3.98; N, 4.81; S, 5.45; Cl, 5.45; Br, 13.5; IR (cm-1)3398, 2652,1671, 1563, 625 and 518 (C-Br). 1H-NMR spectrum (DMSO-d6): δ 1.19, 2.49 (s, 6H, 2x COCH3), δ 3.32(d, 2H, CH2), 4.29 (t, 1H, CH), 5.37, 5.38(d, 1H, CH), 5.40, 5.41 (d, 1H, CH), 7.23-7.64 (m, 9H, Ar–H), 9.05 (s, 1H, SH), 9.99 (s, 1H, OH).18b as white crystals, in 75% yield, m.p. 154°C. Requires: C, 53.57; H, 4.28; N, 5.00; S, 5.71; Br, 14.3; Found: C, 53.87; H, 4.31; N, 5.01; S, 5.82; Br, 14.4; IR (cm-1): 3394, 2644, 1657, 1569, 549 (C-Br). 1H-NMR (DMSO-d6): δ 1.01, 2.49 (s, 6H, 2x COCH3), 3.43 (d, 2H, CH2), δ 4.29 (d, 2H, CH2), 5.39 (t, 1H, CH), 7.06-7.96 (m, 8H, Ar–H), 9.09 (s, 1H, SH), 10.10 (s, 1H, OH). MS (m/z, %): 424 M+2 (0.02%). Synthesis of nucleoside derivatives (19a-c).A suspension of pyrimidin-2-thione derivatives (10b,d,e) (0.01 mol) in 6ml of aqueous KOH solution (prepared from dissolving (0.01 mol) solid KOH in 6ml of distilled water) was well stirred (magnetic stirrer) at room temperature for 3 h, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring. Stirring was continued for further 12 h. After evaporation of the excess solvent (reduced pressure), the residue left was washed with dilute alcohol (several times) and the precipitate formed was recrystallized from ethanol to give (19a-c).19a, as Grey crystals, in 70% yield, m.p. 120°C. Requires: C, 54.64; H, 4.01; N, 5.10; S, 5.82; Cl; 12.9; Found: C, 50.72; H, 4.21; N, 5.21; S, 5.95; Br, 13.1; IR (cm-1): 3294, 1674, 1552,1256, 728 (C-Cl). 1H-NMR (DMSO-d6): δ 2.49, 2.50 (d, 2H, CH2), 3.33, 3.44, 3.59 (t, 1H-CH), 3.71, 4.33 (d, 1H, CH1'–CH2'), 5.380, 5.385 (d, 1H, CH2'), 5.39, 5.40 (d,1H, CH3'), 5.44, 5.45, 5.46 (t, 1H, CH4'), 5.47, 6.65 (d, 2H, CH25'), 6.82-7.73 (m, 8H, Ar–H), and at 10.06 (s, 1H, OH). MS (m/z, %): 478 (0.55%).19b as orange crystals, in 75% yield, m.p. 190°C. IR (cm-1): 3401, 1670, 1595, 1240, 752 (C-Cl). 1H-NMR (DMSO-d6): δ 2.49, 2.50 (d, 2H, CH2), 3.43 (t, 1H, CH), 6.76, 6.77 (d, 1H, CH25'), 6.99-7.61 (m, 7H, Ar–H), 8.18(s, 1H, OH). MS (m/z, %): 480 M+1 (0.22%). 19c as black crystals, 60% yield, m.p. 224°C. Requires: C, 54.71; H, 4.16; N, 5.55; S, 6.34; Cl, 7.03; Found C, 54.89; H, 4.32; N, 5.76; S, 6.56; Cl, 7.12; IR (cm-1): 3343-3225, 1740, 1602, 1243. 1H-NMR (DMSO-d6): δ 3.55 (s, 3H, OCH3), 5.93 (s, 1H, NHCO), 7.01-7.90 (m, 13H, Ar–H), 10.1 (s, 1H, OH). MS (m/z, %): 300 M+2 (0.59%).Synthesis of (2R,3S,4R,5S)-2(4(-2-chlorophenyl)-6-pheny l-4'-1,3-oxazine-2-ylamino)-5'-hydroxytetrahydro-2H-pyran-3',4'-diyl diacetate (20a) and (2R,3S,4R,5S)-2(6-(4-benzamidophenyl)-4-(4-methoxyphenyl)-4H-1,3-oxazin-2-ylamino)-5'-hydroxytetrahydro-2H-pyran-3',4'-diyl diacetate (20b).A suspension of the Oxazine derivatives 11a and/or 11b (0.01 mol) in 6ml of aqueous KOH solution (prepared from dissolving (0.01 mol) solid KOH in 6ml of distilled water) was well stirred (magnetic stirrer) at room temperature for 3 h, then a solution of 12 (0.01 mol) dissolved in 30ml of dry acetone was added drop-wise while stirring. Stirring was continued for further 12 h. After evaporation of the excess solvent (reduced pressure), the residue left was washed with dilute alcohol (several times) and the precipitate formed was recrystallized from ethanol to give 20a,b.20a as pale yellow crystals, 60% yield, m.p. 158-160°C. Requires: C, 64.39; H, 5.36; N, 6.82; Found: C, 64.94; H, 5.40; N, 6.8. IR (cm- 1): 3482, 3302, 1654, 1666, 1591. 1H-NMR (DMSO-d6): δ 2.49, 2.495 (s, 6H, 2xCOCH3), 3.91 (s, 2H, CH2), 4.0 (d, 2H, CH – CH), 4.06 (s, 1H, NH), 7.42 – 7.82 (m, 9H, Ar-H), 8.05 (s, 1H, OH). 20b as yellow crystals, 60% yield, m.p. 110-112°C. Requires: C, 59.94; H, 4.99; N, 5.59; Cl, 7.09; Found: C, 60.1; H, 5.0; N, 5.6; Cl, 7.1. IR (cm-1): 3482, 3302, 1654, 1666, 1591, 1593. 1H-NMR (DMSO-d6): δ 6.9 d, 2H-CH-CH) 2.1 - 2.6 (s, 6H, 2xCOCH3), 10.2 (s, 1H, NH), 10.5 (s, 1H, OH), 3.8 (s, 3H, OCH3), and 7.02 – 8.13 (m, 13H-Ar). 13C-NMR (DMSO-d6): δ 24.1, 26.4, 38.6, 55.3.