Redha I. Al-Bayati1, Mohammed R. Ahamad2, Luma S. Ahamed2
1Department of Chemistry, College of Science, Al-mustansirya University, Baghdad, Iraq
2Department of Chemistry, College of Science, Baghdad University, Baghdad, Iraq
Correspondence to: Luma S. Ahamed, Department of Chemistry, College of Science, Baghdad University, Baghdad, Iraq.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Abstract
New quinoline-2-one derivatives were synthesized by reaction of 4,7-dimethyl coumarin (I) with nitric acid in the presence of concentrated sulfuric acid to afford 4,7-dimethyl-6-nitrocoumarin (II) and 4,7-dimethyl-8-nitrocoumarin (III). The compound (II) was treated with hydrazine hydrate (80% in pyridine) to give N-amino-4,7-dimethy- 6-nitroquinoline-2-one (IV). The latter was used to synthesize 1-(4,7-dimethyl-6-nitro-2-oxo-2H-quinolin-1-yl) -3-phenyl-urea (V) and 1-(4,7-dimethyl-6-nitro-2-oxo-2H-quinolin-1-yl)-3-phenyl-thiourea (VI) by the reaction of (IV) with phenyl isocyanate and phenyl isothiocyanate, respectively. Cyclization of compounds (V) and (VI) with p-phenyl phenacyl bromide gave the corresponding oxazole (VII) and thiazole (VIII), respectively. The diacetylamino derivative (IX) and mono benzoylamino derivative (X) were prepared by reaction of compound (IV) with acetic anhydride and benzoyl chloride, respectively. Subsequently, compound (IV) was reacted with salicyaldehyde to synthesize the corresponding Schiff base (XI). The synthesized compounds were characterized by FTIR, H-NMR, 13C-NMR and by measurements some of their physical properties. Finally, the prepared compounds were evaluated for biological activities against two types of bacteria.
Keywords:
Quinoline-2-One, Hydrazine hydrate, Phenyl isocyanate, Acetyl chloride, Benzoyl chloride, Schiff base
Cite this paper: Redha I. Al-Bayati, Mohammed R. Ahamad, Luma S. Ahamed, Synthesis and Biological Activity Investigation of Some Quinoline-2-One Derivatives, American Journal of Organic Chemistry, Vol. 5 No. 4, 2015, pp. 125-135. doi: 10.5923/j.ajoc.20150504.03.
Article Outline
- 1. Introduction
- 2. Experimental
- 2.1. Instruments
- 2.2. Chemicals
- 2.3. Synthesis of 4,7-dimethylcoumarin (I) [10]
- 2.4. Synthesis of 4,7-dimethyl-6-nitrocoumarin and 4,7-dimethyl-8-nitrocoumarin: (II and III)
- 2.5. Synthesis of 1-amino-4,7-dimethyl-6-nitro-1H- quinoline-2-one: (IV)
- 2.6. Synthesis of 1-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-3-phenylurea (V)
- 2.7. Synthesis of 1-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-3-phenyl-thiourea (V1)
- 2.8. Synthesis of 1-(5-biphenyl-4-yl-5H-oxazol-2-ylideneamino)-4,7-dimethyl-6-nitro -1H-quinolin-2-one (VII)
- 2.9. Synthesis of 1-(5-biphenyl-4-yl-5H-thiazol-2-ylideneamino)-4,7-dimethyl-6-nitro -1H-quinolin-2-one (VIII)
- 2.10. Synthesis of N-acetyl-N-(4,7-dimethyl-6-nitro-2-oxo -2H-quinolin-1-yl)-acetamide (IX)
- 2.11. Synthesis of N-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-benzamide (X)
- 2.12. Synthesis of 1-[(2-hydroxy-benzylidene)-amino]-4,7-dimethyl-6-nitro -1H-quinolin-2-one (XI)
- 2.13. Synthesis of 2-cyano-N-(4,7-dimethyl-6-nitro-2- oxo-2H-quinolin-1-yl)-acetamide (XII)
- 2.14. Antimicrobial Activity Testing
- 3. Results and Discussion
- 3.1. Synthesis and Characterization
- 3.2. Biological Activity
- 4. Conclusions
1. Introduction
Heterocyclic chemistry is one of the largest areas of research in organic chemistry and is growing rapidly due to different types of pharmacological properties [1-2] that heterocycles demonstrate. Coumarin derivatives belong to a group of compounds known as the benzopyrones, all of which consist of a benzene ring joined to a pyranone ring. Coumarin and its derivatives are responsible for high levels of various biological activities [3-5] such as antibacterial, antioxidant, antifungal, antiviral and anticancer activity. The reaction of coumarin with hydrazine hydrate in the presence of pyridine gives N-amino-2-quinolone and its derivatives [6]. Quinolones (carbostyrils or 1-azacoumarins) are among the most popular N-heteroaromatic compounds that are isosteric with coumarins and isomeric to 4-quinolone; these compounds would likely be potential candidates for antibacterial activity. 2-Quinolone derivatives have been shown to have antitumor, anti-inflammatory, antiplatelet, antiulcer, antioxidant and antidepressant biological activities. Many substituted quinolin-2-one derivatives have recently created great interest in chemotherapy as antitumor drugs [7-9]. These biological data prompted us to synthesis some new substituted -1H-2-quinolone derivatives as shown in Scheme 1.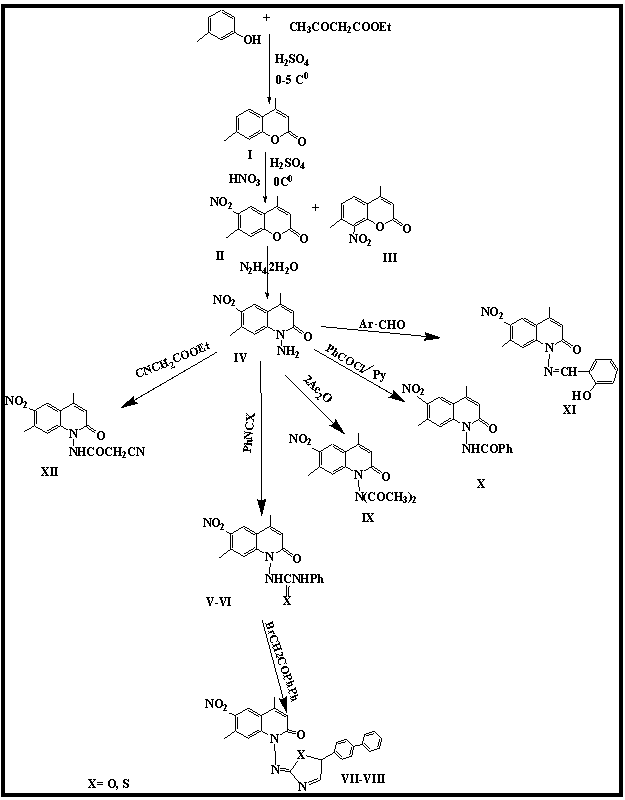 | Scheme 1. Synthesis of some quinoline-2-one derivatives |
2. Experimental
2.1. Instruments
The FTIR spectra in the range (4000–400) cm-1 were recorded without KBr disc on a Shimadzu FTIR 8300 Spectrophotometer. 1HNMR and 13C NMR spectrum (solvent DMSO) was recorded on a Bruker-DPX 400 MHz spectrometer with TMS as internal standard in Isfahan University and on a 300 MHz spectrometer with TMS as internal standard in chemistry department Al-Albayat University, Jordan. Melting points were determined on a Gallen-kamp MFB-600 melting point apparatus and are uncorrected. Analytical thin layer chromatography (TLC) in (7:3 ratio of hexane: ethyl acetate) as the mobile phase was performed on plates precoated with silica gel (Merck 60 F254, 0.25 mm) and was visualized with ultraviolet light.
2.2. Chemicals
Starting chemical compounds were obtained from BDH, Sigma Aldrich and Fluka and were used as received without further purification.
2.3. Synthesis of 4,7-dimethylcoumarin (I) [10]
A mixture of m-cresol (12.18ml, 0.1 mole) and ethyl acetoacetate (12.634ml, 0.1 mole) was cooled to 0°C, concentrated sulfuric acid 46ml was added drop wise in such a way that the temperature does not rise above 8°C. After that, the stirring was continued for one hour at room temperature. Then the mixture was heated at 60-70°C for 6 hrs, after cooling, the solution was poured into ice /cold water. The solid product was filtered, and washed with cold water, dried at room temperature, then recrystallized from mixture EtOH:H2O (1:1) as white crystals, yield 80%, m.p. 131-133°C, Rf=0.6. IR and NMR data: see Tables 1-3.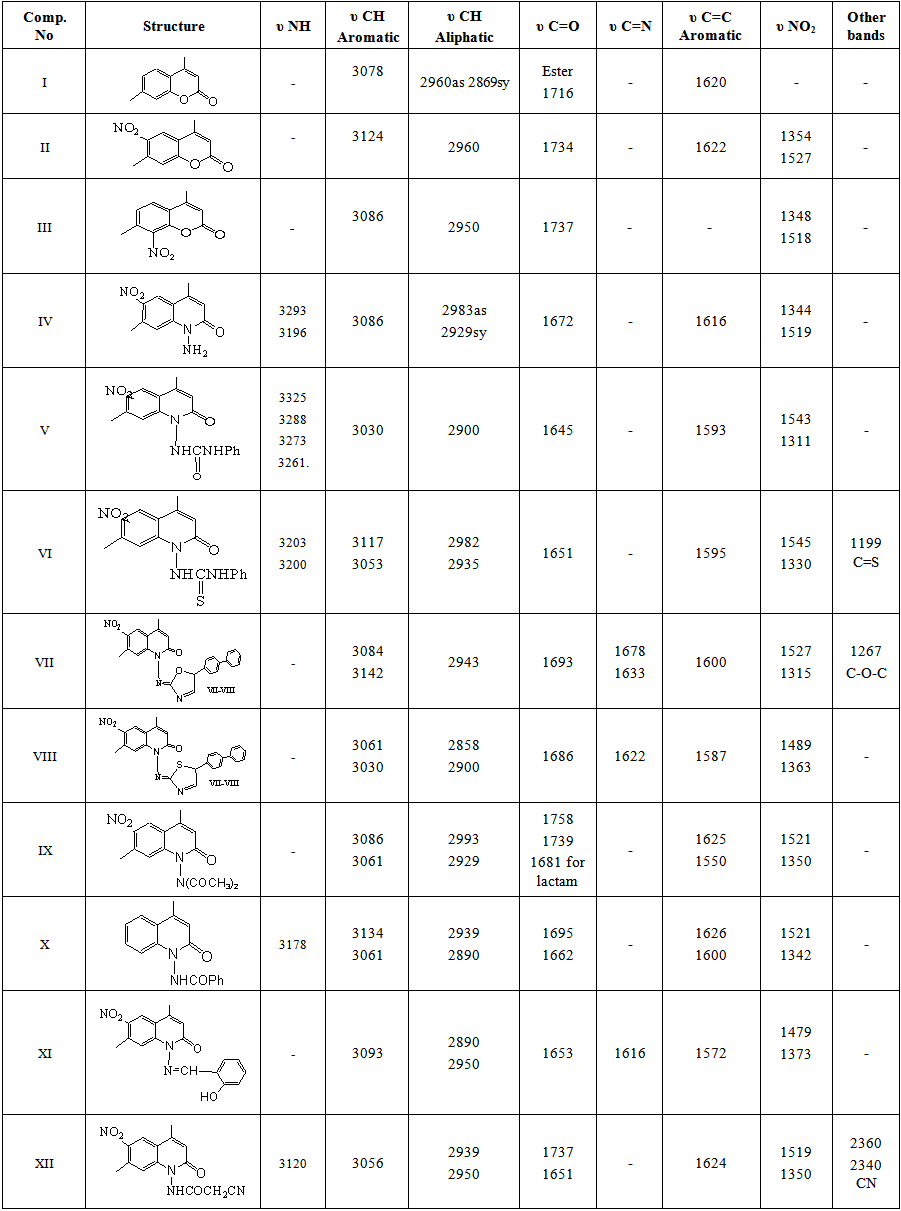 | Table 1. FTIR data of synthesized compounds I-XIV |
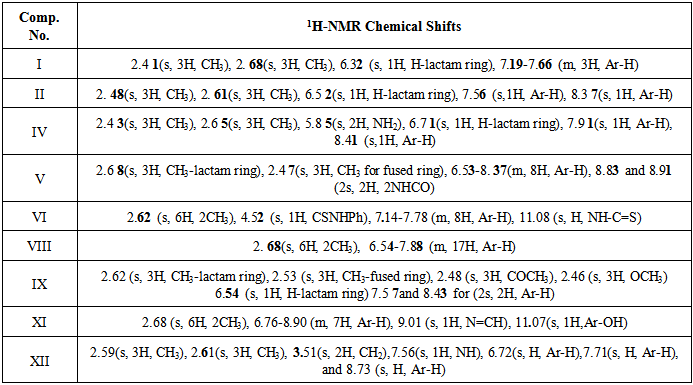
Table 2. 1H-NMR of some synthesized compounds
 |
| |
|
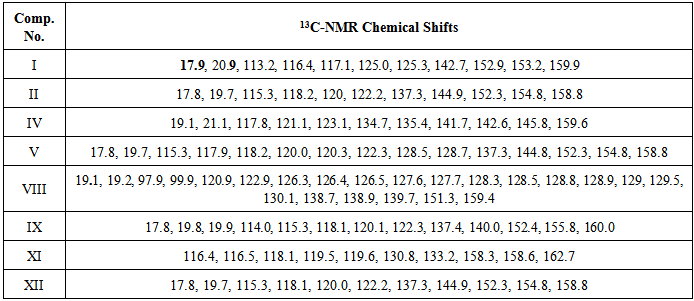
Table 3. 13C-NMR of some synthesized compounds
 |
| |
|
2.4. Synthesis of 4,7-dimethyl-6-nitrocoumarin and 4,7-dimethyl-8-nitrocoumarin: (II and III)
Prepared according to the published procedures [11] and [12] with some modifications:A mixture of 4,7-dimethylcoumarin I (0.696 gm, 0.004 mole), and conc. sulfuric acid (75ml) was stirred at 0°C for 20 min. Then, a mixture of conc. sulfuric acid (1.25ml, 98%), and conc. nitric acid (0.4ml, d1.4) was added at (0-5)°C and the reaction mixture was stirred for 3 hrs at 5°C. Then, it was poured into an ice/cold water solution, the solid product was filtered and washed with cold water, dried at room temperature, and the crude mixture was separated in ethanol by using a soxhlet apparatus. The precipitate that did not dissolve in hot ethanol was 4,7-dimethy-6-nitrocoumarin (II) which was recrystallized from ethyl acetate as pale yellow crystals, yield 80%, m.p. 259-260°C, Rf= 0.5. IR and NMR data: see Tables 1-3.The filtrate was concentrated and cooled, yielding a precipitate, 4,7-dimethyl-8-nitrocoumarin (III) which was recrystallized from ethyl acetate in 20% yield, m. p. 242-243°C, Rf= 0.4. IR data: See Table 1.
2.5. Synthesis of 1-amino-4,7-dimethyl-6-nitro-1H- quinoline-2-one: (IV)
Compound II (10.95gm, 0.05mole) was dissolved in anhydrous pyridine (20ml) and hydrazine hydrate (80%, 0.15mole) was added to the mixture. The reaction mixture was refluxed for 6 h with stirring at 117°C. The resulting solution was concentrated under reduced pressure, and additional ethanol (10ml) was added. The excess solvent was concentrated under reduced pressure again. The crude product was recrystallized from ethanol or toluene as yellow crystals, yield 95%, m.p. 269-271°C, Rf= 0.16, IR and NMR data: See Tables 1-3.
2.6. Synthesis of 1-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-3-phenylurea (V)
Phenyl isocyanate (0.648ml, 0.006mole) was added to a solution of compound IV (1.4g, 0.006mole) in absolute ethanol (20ml) and refluxed for 24 h. The solution was concentrated at room temperature. Then, diethyl ether was added to the residue and the precipitate was filtered immediately. The solid crude product was recrystallised from a mixture of ethanol and water, affording yellow crystals in 75% yield, m.p. 240-245°C, Rf= 0.53. IR and NMR data: see Tables 1-3.
2.7. Synthesis of 1-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-3-phenyl-thiourea (V1)
Phenyl thiocyanate (0.72ml, 0.006mol) was added to a solution of compound IV (1.4g, 0.006mole) in absolute ethanol (20ml) and refluxed for 24hr. The solution was concentrated and poured into ice water. The precipitate was filtered and recrystallized from EtOH with water, affording yellow needle-like crystals in 73% yield, m.p. 50-55°C, Rf= 0.82. IR and NMR data: see Tables 1 and 2.
2.8. Synthesis of 1-(5-biphenyl-4-yl-5H-oxazol-2-ylideneamino)-4,7-dimethyl-6-nitro -1H-quinolin-2-one (VII)
Compound V (0.627g, 0.0017mole) was dissolved in absolute ethanol (15ml) and p-phenyl phenacylbromide (0.47g, 0.0017mole) was added to the solution, refluxed for 18 h, then cooled and neutralized with ammonium hydroxide solution. The precipitate was washed with petroleum ether (60-80°C) twice, then with water and a mixture of acetone: water was used for recrystallization to give a yellow powder in a yield of 80%, m.p. 230-231°C, Rf= 0.72. IR data: see Table 1.
2.9. Synthesis of 1-(5-biphenyl-4-yl-5H-thiazol-2-ylideneamino)-4,7-dimethyl-6-nitro -1H-quinolin-2-one (VIII)
Compound VI (0.6273g, 0.0017mole) was dissolved in absolute ethanol (15ml) and p-phenyl phenacylbromide (0.47g, 0.0017mole) was added to the solution, refluxed for 16 h, then cooled and neutralized with ammonium hydroxide solution. Cold water was added to a solution to complete the precipitation. The precipitate was filtered off, washed with water, and a mixture of ethanol/water was used for recrystallization to give a yellow powder in 85% yield, m.p. 215-217°C dec., Rf= 0.7. IR and NMR data: see Tables 1-3.
2.10. Synthesis of N-acetyl-N-(4,7-dimethyl-6-nitro-2-oxo -2H-quinolin-1-yl)-acetamide (IX)
A mixture of compound IV (0.583gm, 0.0025 mole) and acetic anhydride (5ml) was heated under reflux for 5 h. Then, the reaction mixture was cooled and poured into ice. The solid product was filtered off, washed with water, dried and recrystallized from acetone/water as a brown powder, yield 75%, m.p. 256-258°C, Rf= 0.45. IR and NMR data: see Tables 1-3.
2.11. Synthesis of N-(4,7-dimethyl-6-nitro-2-oxo-2H- quinolin-1-yl)-benzamide (X)
A mixture of IV (0.583gm, 0.0025mole) and benzoyl chloride (5ml) in 30 ml of anhydrous pyridine was refluxed for 4hr. The reaction mixture was cooled to room temperature. The obtained product was recrystallized from acetone/water as brown crystals, yield 70%, m.p. 242-245°C, Rf= 0.55. IR data: See Table 1.
2.12. Synthesis of 1-[(2-hydroxy-benzylidene)-amino]-4,7-dimethyl-6-nitro -1H-quinolin-2-one (XI)
A mixture of salicyaldehyde (1.6ml, 0.015mole) and IV (3.5gm, 0.015mole) in 25ml absolute ethanol in the presence of 4 drops of glacial acetic acid was refluxed for 8 h. The progress of the reaction was monitored by TLC (7:3 hexane:ethyl acetate). Upon completion of the reaction, the mixture was cooled and poured into an ice/cold water mixture. The obtained product was filtered and recrystallized from a 1:1 mixture of EtOH:H2O, affording a pale yellow powder, yield 71%, m.p. 216°C dec., Rf= 0.74. IR and NMR data: See Tables 1-3.
2.13. Synthesis of 2-cyano-N-(4,7-dimethyl-6-nitro-2- oxo-2H-quinolin-1-yl)-acetamide (XII)
Ethyl cyanoacetate (0.32ml, 0.003mole) was added to a solution of IV (0.699g, 0.003mole) in DMF (20ml). Then, a small amount of p-toluenesulphonic acid was added and refluxed for 10 h. The resulting reaction mixture was poured into an ice/water solution. The solid product was filtered off and recrystallized from EtOH:H2O as a brown powder, yield 67%, m.p. 260°C Dec., Rf= 0.68. IR and NMR data: See Tables 1-3.
2.14. Antimicrobial Activity Testing
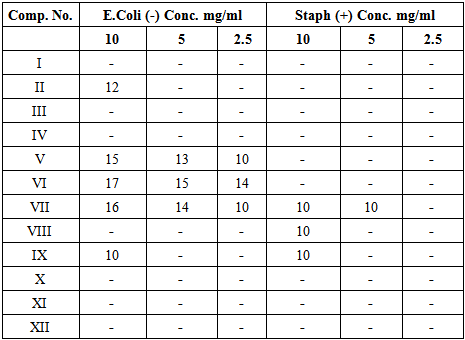
The synthesized compounds were prepared with different concentrations (10, 5, and 2.5 mg/ml) using dimethyl sulfoxide (DMSO) as solvent. The agar well diffusion method was used to determine antimicrobial activity. The culture medium was inoculated with two types of bacteria (E. Coli and staphylococcus aureus) suspended in nutrient agar plates. Six millimeter diameter wells punched into the agar with fresh bacteria and filled with (100 μL) of each concentration. DMSO was used as a control. The incubation was carried out at 37°C for 24 h. Solvent and growth controls were kept and zones of inhibition were measured. The antimicrobial activity was evaluated by measuring the inhibition zone diameter observed was recorded. All data were summarized in the Table 4.Table 4. Biological activity of synthesized compounds. (Inhibition zone measurements in mm)
 |
| |
|
3. Results and Discussion
3.1. Synthesis and Characterization
4,7-dimethylcoumarin (I) was synthesized by treatment of m-cresol with methyl acetoacetate in the presence of conc. sulfuric acid under Pechmann condensation conditions. The FTIR of compound (I) showed absorption bands at 1716 cm-1 for (υ C=O) of the lactone ring. The 1HNMR spectrum showed signals at 2.48 and 2.6 ppm due to two CH3 groups [13], a singlet signal at 6.3 ppm for one proton of the lactone ring and signals at 7.19 -7.66 ppm due to three aromatic protons. The 13C-NMR showed two signals at 17.9 and 20.9 ppm due to two CH3 groups and a signal at 159.9 due to the C=O of the lactone ring. The nitration of compound I by nitric acid in the presence of sulfuric acid at 0°C through an electrophilic aromatic substitution mechanism gave a mixture of 80% 4,7-dimethyl-6-nitrocoumarin II m.p. 259-260°C, and 20% 4,7-dimethyl-8-nitrocoumarin III m.p 242-243°C. The FTIR spectrum of compound II showed the appearance of new two absorption bands at 1527 and 1354 cm-1 belonging to the asymmetric stretch and symmetric stretch of NO2 group, respectively[14]. The 1H-NMR spectrum of compound II showed signals at 2.48 ppm and 2.61 ppm for two methyl groups, a signal at 6.52 ppm due to the proton of the lactone ring and two signals at 7.56 and 8.37 ppm due to two protons of the aromatic ring. The 13C-NMR spectrum showed signals at 17.8 and 19.7 ppm for two CH3 groups, 154.8 ppm (C-O), 152.3ppm (C-NO2), and signals ranging from 115.3-122.2 ppm for three aromatic ring carbons and 158.8 ppm for the carbonyl carbon of the lactone ring (O-C=O). See Figures 1 and 2.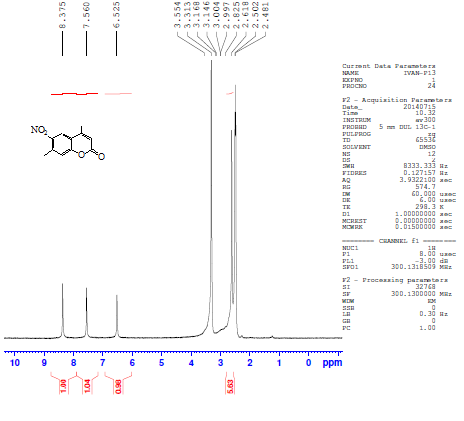 | Figure 1. 1H-NMR spectrum of compound II |
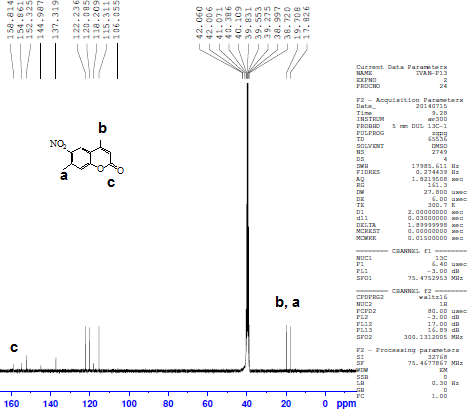 | Figure 2. 13C-NMR spectrum of compound II |
Another isomeric nitrocoumarin derivative was 4,7-dimethyl-8-nitrocoumarin III which showed different physical properties like solubility in hot ethanol (very soluble), melting point and Rf of TLC from compound II. The FTIR of compound III showed the strong band at 1737 cm-1 for carbonyl group and two strong bands at 1518 and 1348 cm-1 for the asymmetric stretch and symmetric stretch of the NO2 group. The treatment of compound II with excess hydrazine hydrate produced N-aminoquinoline-2-one derivative IV. This reaction proceeded by nucleophilic addition of hydrazine to the carbonyl group of a cyclic ester (lactone) giving the corresponding amino compound IV. The FTIR spectrum of compound IV showed the appearance of amine absorption bands at 3293 and 3196 cm-1 and 1672 cm-1 due to υC=O amid of cyclic lactam group Table 1. The 1H-NMR spectrum of compound IV showed signals at 2.41 ppm and 2.68 ppm for two methyl groups (CH3), a signal at 5.85 ppm for the two protons of NH2, a signal at 6.71 ppm for the proton of the lactam ring, and signals at 7.91 and 8.41 ppm for two aromatic protons. See Figure 3. The 13C-NMR spectrum of compound IV showed signals at 18.5 and 20.5 ppm for two methyl groups (2CH3) and showed signals at 117.6, 134.7, 135.4, and 141.8 ppm for aromatic carbons, 142.6 ppm for the amino group carbon C-N-NH2, 145.6 ppm for C-NO2 and 159.6 ppm for the carbonyl carbon of the lactam ring. See Figure 4.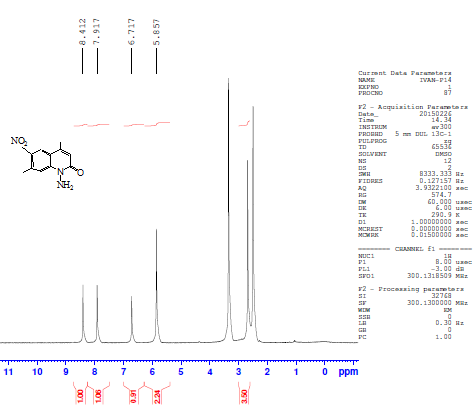 | Figure 3. 1H-NMR spectrum of compound (IV) |
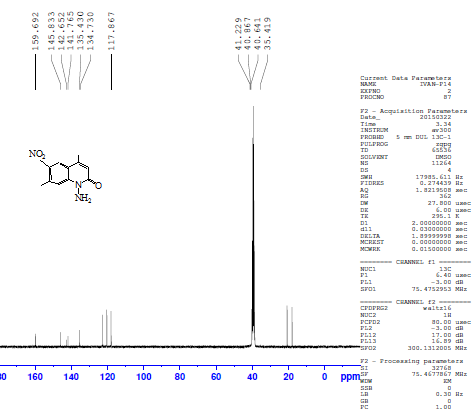 | Figure 4. 13C-NMR spectrum of compound (IV) |
The treatment of compound IV with phenyl isocyanate and phenyl isothiocyanate in ethanol afforded compounds V and VI through the nucleophilic addition of the amine to the carbon of the isocyanate group or carbon of the isothiocyanate group. The FTIR spectra of compounds V, and VI showed the disappearance of absorption bands of the υNH2 at 3293 and 3196 cm-1, and the appearance of new absorption bands for υNH at 3325-3200 cm-1, and new absorption band at 1645 cm-1 for υC=O (imide) of compound V and at 1199 cm-1 for υC=S (thiourea) for compound VI. The 1H-NMR spectrum of the compound V showed new signals at 8.83 and 8.91 ppm for two protons of amide groups (2 NHC=O). The intramolecular cyclization of compound V with phenyl phenacyl bromide led to formation of the oxazole derivative VII. The FTIR spectrum of compound VII showed an absence of absorption bands for two (υNH) and presence of new υC=N absorption bands at 1678 and 1633 cm-1 for the imino groups. The intramolecular cyclization of compound VI with phenyl phenacyl bromide led to the thiazole derivative VIII. The FTIR spectrum of compound VIII showed a disappearance of absorption bands υNH and υC=S groups and showed a new absorption band at 1622 cm-1 due to υC=N. The 1H-NMR spectrum of compound VIII showed a multiplet signal at 6.54-7.88 ppm which we ascribe to the aromatic protons. The 13C-NMR spectrum showed a new signal 99.9 ppm for the olefinic C-H. See Table 3.The diacetylation of amino derivatives was achieved by the reaction of a compound IV with the excess amount of acetic anhydride without using any solvent or catalyst to afford the diacetyl derivative IX. The FTIR showed disappearance of the υNH2 group absorption and showed new absorption bands at 1758 and 1739 cm-1 due to two υC=O of the two acetyl groups in addition to 1681 cm-1 due to υC=O of the lactam ring. The high frequencies of two amide carbonyl groups may due to the presence of the two carbonyl groups on the amine, which would serve to decrease the resonance stabilization normally observed in amides Table 1. The 1H-NMR spectrum (Figure 5) showed two singlets at 2.48 and 2.46 ppm for the protons of the acetyl groups (2 COCH3) [12]. The 13C-NMR showed a new signal at 19.9 ppm due to the methyl carbons of the diacetyl groups (COCH3). See Tables 2 and 3. The Rf of compound IX was 0.45 in a 3:7 mixture of ethyl acetate:hexane while the Rf for compound IV was 0.16 in the same eluent. When compound IV was treated with benzyl chloride in anhydrous pyridine as a solvent and catalyst, derivative X was obtained in good yield. The FTIR Spectrum of compound X indicated the absence of absorption bands for υNH2 and presence of absorption band at 3178 cm-1for υNH band and presence of absorption band at 1695 and 1662 cm-1 for the υC=O of the benzoyl group and lactam ring, respectively.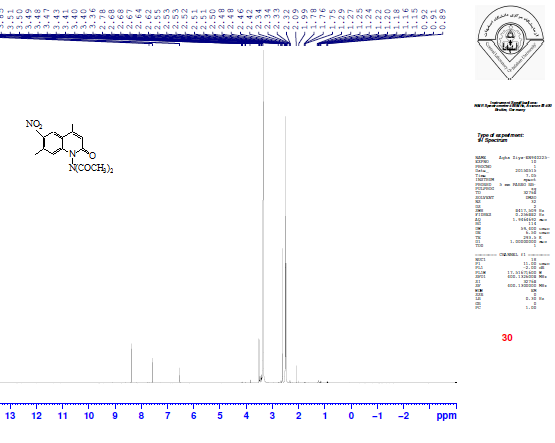 | Figure 5. 1H-NMR spectrum of compound IX |
The formation of the Schiff base 1-[(2-Hydroxybenzylidene)-amino]-4,7-dimethyl-6-nitro-1H-quinolin-2-one XI was confirmed by the absence of absorption band for υNH2 and presence of a new absorption band at 1616 cm-1 due to υC=N starching. The 1HNMR spectrum of compound XI showed a new signal observed at 9.01 ppm integrating for (CH=N), signals at 6.76-8.9 ppm integrating for aromatic protons of the aryl groups and at 11.07 ppm for the proton of the phenolic OH group as shown in Figure 6.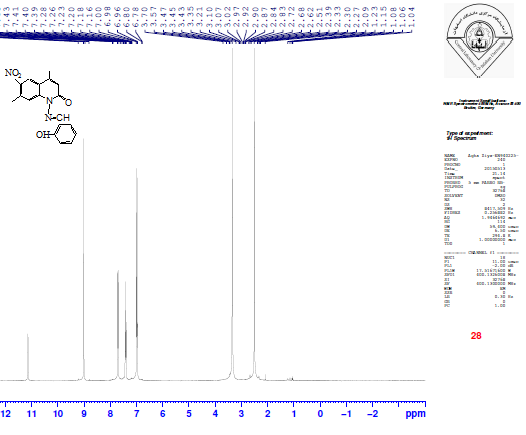 | Figure 6. 1H-NMR spectrum of compound XI |
The reaction of IV with ethyl cyanoacetate gave 2-cyano-N-(4,7-dimethyl-6-nitro-2-oxo-2H-quinolin-1-yl)-acetamide XII. The FTIR spectrum of compound XII revealed CN stretching at 2340 cm-1, the absence of absorption band for υNH2 and a new characteristic υC=O stretching of the amide at 1737cm-1, the high frequency likely due to the electron-withdrawing nature of the CN group. See Table 1. Moreover, the 1H-NMR spectrum (Figure 7) exhibited a singlet at 3.51 ppm for two protons of the acetamido CH2, a singlet at 7.56 ppm for acetamido N-H proton and three singlet signals at 6.72, 7.71 and 8.73 ppm for three aromatic protons. See Tables 2 and 3.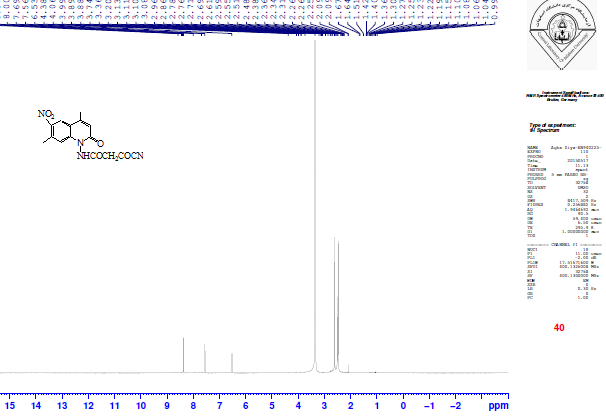 | Figure 7. 1H-NMR spectrum of compound XII |
3.2. Biological Activity
The synthesized compounds were preliminary evaluated for their antibacterial activity against gram-negative bacteria, E. Coli, and gram-positive bacteria, S. aureus, using the disk diffusion method. The recorded inhibition zones were measured in millimeters and summarized in Table 4. We observed some results from the data of inhibition zone: The compounds V-VII showed the most activity against E. Coli, while they were found to be inactive or less active toward S. aureus. Compound VII demonstrated the broadest band of activity against of the two types of bacteria. This could be related to a synergistic effect of having oxazole ring and imine linkage together in the structure. Compound VII (thiourea derivative) was determined to be more active than compound V (urea derivative) against E.Coli finding that may be related to the present of the thiourea moiety. These preliminary screening test results are promising and warrant further studies.
4. Conclusions
In conclusion, several new quinoline -2-one derivatives were synthesized via a new method for separating two isomers II and III by incorporation of a soxhlet apparatus, a novel experimental route was developed and used for isolation of a new compound IV which was prepared in good yield. Compound IV was then converted into a variety of derivatives. Moreover, several of the newly synthesized compounds showed antimicrobial activity against E. coli rather than Staphylococcus aureus, while compound VII exhibited a broad spectrum of biological activity against both E.coli and staphylococcus aureus.
References
| [1] | Kawkab, Y. S., AL-Bayati, R. I., and Mohammed, K. H. “Synthesis of New coumarin and 2-quinolone Derivatives With Expected Biological Activities,” Iraqi J Pharm Sci, Vol. 21(II):42-49 (2012). |
| [2] | Ramasamy, A. K., Balasubramaniam, V. and Mohan, k. “Synthesis and Characterization of substituted 4-Methoxy-1H-Quinolin-2-ones,” E-Journal of Chemistry, 7(III): 1066-1070(2010). |
| [3] | Hament, P., Nidhi, C., Ranjana, D., and Tilak, R. “Quinoline-6-Amines: Synthesis and Biological evaluation,” Indo. J. Chem., 13(III): 185-192(2013). |
| [4] | Stoyanov, E., Ivanov, C. I. “Convenient Replacement of the Hydroxy by an Amino Group in 4-Hydroxycoumarin and 4-Hydroxy-6-methyl-2-Pyrone under Microwave Irradiation,” Molecules, 9:627-631 (2004). |
| [5] | Sanjay, K., Anil, S., and Jagir, S. S. “LiBr-Mediated, solvent free von Pechmann reaction: facile and efficient method for the synthesis of 2H-chromen-2-ones,” ARKIVOC, (xv), 18-23 (2007). |
| [6] | AL-Bayati, R., Mahdi, F. “Synthesis of Novel 2-Quinolone Derivatives,” African J. Pure and Appl. Chem., 4X:228-232 (2010). |
| [7] | Abhishek, K., Jennifer, F., Pankaj, K.” Synthesis, Antimicrobial and Anti-inflammatory Activity of Newly Synthesized Isoxazoline Incorporated 2-Quinolones,” International Journal of Pharmaceutical Sciences and Drug Research, 6(2): 124-127 (2014). |
| [8] | Pawar, P. Y., Mane,, B. Y., Salve, M. T. and Bafana, S. R. “Synthesis and anticonvulsant activity ofN-Substituted-7-hydroxy-4-methyl-2-oxa-quinoline derivatives,” International Journal of Drug Research and Technology, 3(3):60-66 (2013). |
| [9] | Abd Al-Jabbar, A. A. “Synthesis and Characterization of New Compounds of Quinolin -2-ones and Evaluation of Biological Activity of Some of Them,” Ph.D. Thesis. Department of Chemistry, College of science, University of Tikrite, Iraq. (2010). |
| [10] | Abolfath, P., Ali, K., Saghar, M. H., Rahele, B., Abdolkarim, Z., Ahmad, R. M., and Marzieh, N. “Silica supported boric tri-sulfuric anhydride as a novel and efficient catalyst for solvent-free synthesis of coumarins via Pechmann condensation,” ARKIVOC, (ix): 111-121 (2012). |
| [11] | Nofal, Z. M., El-Zahar, M. I., and Abd El-Karim, S. S. “Novel coumarin Derivatives with Expected Biological Activity,” Molecules, 5:99-113 (2000). |
| [12] | Swayam S. S.,and Smita S. N. et al. “Synthesis of novel coumarin derivatives and its biological evaluations,” Euro. J. Exp. Bio., 2(4): 899-908 (2012). |
| [13] | Kiran, K. P., Anilkumar, G. N., Manohar, V. K., “Synthesis, Spectroscopic and Crystal Structure Studies on Ethyl-5, 7-Dimethyl coumarin -4-Acetate,” Scientific Research, 2:88-92 (2013). |
| [14] | Silverstein, R., Webster, F. and Kiemle, D., Spectroscopic identification of organic compounds, 7th Ed., John Wiley & Sons, Inc., USA (2005). |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML