-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Organic Chemistry
p-ISSN: 2163-1271 e-ISSN: 2163-1301
2015; 5(4): 116-124
doi:10.5923/j.ajoc.20150504.02
Design and Synthesis of Two New Epoxides
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFigueroa-Valverde Lauro1, Díaz-Cedillo Francisco2, García-Cervera Elodia1, Rosas-Nexticapa Marcela3, Pool-Gómez Eduardo1, Camacho-Luis Abelardo4, García-Martínez Rolando1, Lopéz-Ramos Maria1, García-Camacho Tania1, Mijangos-Gómez N. Scarlet1
1Laboratory of Pharmaco-Chemistry at the Faculty of Chemical Biological Sciences from the University Autonomous of Campeche, Av. Agustín Melgar s/n, Col Buenavista, Campeche Cam., México
2Esc. Nal. de Ciencias Biológicas del Inst. Pol. Nal. Prol. Carpio y Plan de Ayala s/n Col. Santo Tomas, México
3Facultad de Nutrición, Universidad Veracruzana. Médicos y Odontólogos s/n, Xalapa Veracruz, México
4Fac. de Med. y Nutr., Universidad Juárez del Estado de Durango, Av. Universidad esq. Fanny Anitúa, Durango, México
Correspondence to: Figueroa-Valverde Lauro, Laboratory of Pharmaco-Chemistry at the Faculty of Chemical Biological Sciences from the University Autonomous of Campeche, Av. Agustín Melgar s/n, Col Buenavista, Campeche Cam., México.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
In this study is reported a straightforward route for synthesis of two new epoxides using some strategies. The first stage was achieved by the synthesis of the cyclohexylimino-azetidin-benzoic acid (3) by the reaction of 4-hydroxy benzoic acid with a nitrobenzamide derivative. Following, 3 was reacted with ethylenediamine using boric acid as catalyst to form a phenoxy-carboxamide analog (4). In addition, 4 was used to react with chloroacetyl chloride in presence of triethylamine for preparation of a chloroamide derivative (5). Then, 5 was reacted with 2-hydroxy-1-naphthaldehyde in basic medium for the synthesis of an epoxide-benzamide analog (6). The second stage was developed by the synthesis of an enone-derivative (10) using the three component system (cinnamaldehyde, 4-aminoantipyrine and alkyne-1) in presence of cupric chloride. In addition 10 was reacted with ethylenediamine in presence of boric acid to form an phenylcyclopentamine derivative (11). After, 11 was reacted with chloroacetyl chloride using treiethylamine as catalyst to form a chloroacetamide analog (12). Following, 12 was reacted with 2-hydroxy-1-naphthaldehyde in basic medium for preparation of an epoxide-amide derivative (13). The structure of the compounds obtained was confirmed using elemental analysis and NMR spectra. The proposed method offers some advantages such as simple procedure and ease of workup.
Keywords: Epoxide, Naphthaldehyde, Derivative, Enone
Cite this paper: Figueroa-Valverde Lauro, Díaz-Cedillo Francisco, García-Cervera Elodia, Rosas-Nexticapa Marcela, Pool-Gómez Eduardo, Camacho-Luis Abelardo, García-Martínez Rolando, Lopéz-Ramos Maria, García-Camacho Tania, Mijangos-Gómez N. Scarlet, Design and Synthesis of Two New Epoxides, American Journal of Organic Chemistry, Vol. 5 No. 4, 2015, pp. 116-124. doi: 10.5923/j.ajoc.20150504.02.
1. Introduction
- Since years ago several epoxide derivatives have been prepared using protocols different; for example, the synthesis of1-[(Ethoxycarbonyl)methyl]tetrahydrothiophenium Bromide by the reaction of tetrahydrothiophene with ethyl bromoacetate [1]. Other data showed the preparation of 3-oxyranil-chlorophyll by the reaction of Methyl pyropheophorbide with a chlorophyll derivative [2]. In addition the compound methyl 4-(3-hydroxycyclohex-1-en- 1-yl)buta- noate was reacted with m-chloroperoxybenzoic acid (mCPBA) to form the epoxide derivative (methyl 4-(5-hydroxy-7-oxabicyclo[4.1.0]hept-1-yl)butanoate) [3]. Other study shown the synthesis of 1,1-Bis(2,3-epoxy- cyclohexyloxymethyl)-3,4-epoxycyclohexane by the reaction of1,1-Bis(2-cyclohexenyloxymethyl)-3-cyclohexene (I) with mCPBA [4]. Another report indicate preparation of the compound phenyl [(2S,4R)-4-phenyloxetan-2-yl] methanone by the reaction of(2E)-1,3-bis(4-methoxyphenyl) prop-2-en-1-one with potassium hydroxide [5]. In addition, a study showed the synthesis of a vinyl carbamate-epoxide by reaction of vinyl carbamate with dimethyldioxirane [6]. Recently was synthesized the compound (2R,3S)-N-(3-(5-H- dibenzo[b,f]azepin-5-yl)-2,5-dihydro-1,2,4-thiadiazol-5-yl)-3-phenyloxirane-2-carboxamide by the reaction of N-[3-(5H-diben-zo[b,f]azepin-5-yl)-2,5-dihy-dro-1,2,4-thiadiazol-5-yl] propanamide with sodium hydroxide [7]. All these experimental results show several procedures which are available for synthesis of several epoxide-derivatives; nevertheless, expensive reagents and special conditions are required. Therefore, in this study two new epoxide-derivatives were synthesized using several strategies.
2. Experimental
- General methodsThe compound 1 (N-(3-Butyl-1-cyclohexyl-4-ciclohexyl- imino-azetidin-2-ylidene)-4-nitro-benzamide) was prepared with methods previously reported [8]. In addition, all the reagents used in this study were purchased from Sigma-Aldrich Co. Ltd. The melting points for the different compounds were determined on an Electrothermal (900 model). Infrared spectra (IR) were recorded using KBr pellets on a Perkin Elmer Lambda 40 spectrometer. 1H and 13C NMR and 2D-COSY spectra were recorded on a Varian VXR-300/5 FT NMR spectrometer at 300 and 75.4 MHz in CDCl3 using TMS as internal standard. EIMS spectra were obtained with a Finnigan Trace GCPolaris Q. spectrometer. Elementary analysis data were acquired from a Perkin Elmer Ser. II CHNS/0 2400 elemental analyzer.4-[4-(3-Butyl-1-cyclohexyl-4-cyclohexylimino-azetidin-2-ylidenecarba- moyl)-phenoxy]-benzoic acid (3)A solution of 1 (200 mg, 0.44 mmol), 4-hydroxybenzoic acid (70, 0.50 mmol) potassium carbonate anhydrous (42 mg, 0.30 mmol) in 5 ml of Dimethyl sulfoxide was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (3:1) yielding 80% of product, m.p. 222-224°C; IR (Vmax, cm-1): 1680, 1722 and 1224; 1H NMR (300 MHz, CDCl3) δH: 0.86 (s, 3H), 1.08-1.20 (m,7H), 1.30-1.44 (m, 7H), 1.46 (m, 2H), 1.50-1.54 (m, 3H), 1.58 (t, 2H, J = 6.0 Hz), 1.62-1.66 (m, 3H), 1.80-1.90 (m, 2H), 3.18-3.20 (m, 2H), 4.80 (m, 1H), 6.94 (m, 2H), 7.00-8.10 (m, 4H), 8.44 (m, 2H), 10.80 (broad, 1H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 14.16, 23.00, 23.08, 24.40, 25.36, 26.00, 26.28, 29.10, 29.66, 32.20, 38.70, 57.88, 59.68, 116.70, 117.22, 124.18, 131.00, 132.60, 133.84, 134.46, 157.10, 164.09, 165.08, 168.40, 175.94 ppm. EI-MS m/z: 543.30 (M+ 9). Anal. Calcd. for C33H41N3O4: C, 72.90; H, 7.60; N, 7.73; O, 11.77. Found: C, 72.84; H, 7.55.N-(2-aminoethyl)-4-(4-(((2Z,4Z)-3-butyl-1-cyclohexyl-4-(cyclohexylimino)azetidin-2-ylidene)carbamoyl)pheno- xy)benzamide (4)A solution of 3 (200 mg, 0.37 mmol), ethylenediamine (80 µl, 0.74 mmol), and boric acid (40 mg, 0.60 mmol) in 5 ml of methanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (4:1) yielding 65% of product, m.p. 268-270°C; IR (Vmax, cm-1):3378, 3320 and 1686; 1H NMR (300 MHz, CDCl3) δH: 0.86 (s, 3H), 1.08-1.20 (m, 7H), 1.32-1.44 (m, 7H), 1.46 (m, 2H), 1.50-1.54 (m, 3H), 1.58 (t, 2H, J = 6.96 Hz), 1.60-1.64 (m, 3H), 1.80-1.90 (m, 2H), 3.12 (t, 2H, J = 6.44 Hz), 3.18-3.22 (m, 2H), 3.50 (t, 2H, J = 6.44 Hz), 4.62 (broad, 3H), 4.80 (m, 1H), 6.82 (m, 2H), 6.98 (m, 2H), 7.64 (m, 2H), 8.36 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 14.16, 23.00, 23.10, 24.40, 25.46, 26.00, 26.30, 29.12, 29.68, 32.20, 38.66, 43.28, 43.66, 57.90, 59.68, 116.64, 117.38, 128.700, 130.88, 131.00, 132.60, 134.37, 160.92, 163.21, 164.10, 169.64, 175.88 ppm. EI-MS m/z: 585.36 (M+ 10). Anal. Calcd. for C35H47N5O3: C, 71.76; H, 8.09; N, 11.96; O, 8.19. Found: C, 71.70; H, 8.00.N-((2Z,4Z)-3-butyl-1-cyclohexyl-4-(cyclohexylimino)azetidin-2-ylidene)-4-(4-((2-(2-chlroacetamido)ethyl)carba- moyl)phenoxy)benzamide (5)A solution of 4 (200 mg, 0.34 mmol), triethylamine (100 μl, 1.50 mmol) and chloroacetyl chloride (128 μl, 1.60 mmol) in 5 ml of methanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water:hexane (4:1:2) yielding 44% of product, m.p. 280-282°C; IR (Vmax, cm-1):3380, 1684 and 1226; 1H NMR (300 MHz, CDCl3) δH: 0.88 (s, 3H), 1.08-1.20 (m, 7H), 1.30-1.46 (m, 7H), 1.48 (m, 2H), 1.50-1.54 (m, 3H), 1.58 (t, 2H, J = 6.96 Hz), 1.62-1.66 (m, 3H), 1.80-1.90 (m, 2H), 3.18-3.22 (m, 2H), 3.50 (m, 4H), 4.00 (t, 2H, J = 13.62 Hz), 4.80 (m, 1H), 6.82 (m, 2H), 6.98 (m, 2H), 7.64 (m, 2H), 8.02 (broad, 2H), 8.38 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 14.18, 23.00, 23.10, 24.40, 25.36, 26.00, 26.30, 29.22, 29.67, 32.24, 38.70, 38.80, 38.84, 42.40, 57.88, 59.82, 116.82, 117.34, 128.90, 130.88, 131.00, 132.70, 134.48, 161.04, 162.30, 162.58, 163.32, 164.13, 176.00 ppm. EI-MS m/z: 661.33 (M+ 12). Anal. Calcd. for C37H48ClN5O4: C, 67.10; H, 7.31; Cl, 5.35; N, 10.57; O, 9.66. Found: C, 67.03; H, 7.24.N-((2Z,4Z)-3-butyl-1-cyclohexyl-4-(cyclohexylimino)aze-tidin-2-ylidene)-4-(4-((2-(2-(3-(2-hydroxynaphthalen-1-yl)oxiran-2-yl)acetamido)ethyl)carbamoyl)phenoxy)ben- zamide (6)A solution of 5 (200 mg, 0.30 mmol), 2-hydroxy- 1-naphthaldehyde (68 mg, 0.40 mmol), and sodium hydroxide (20 mg, 0.50 mmol) in 5 ml of ethanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol: water: hexane (4:1:2) yielding 38% of product, m.p. 180-182°C; IR (Vmax, cm-1):3380, 3222, 1680, and 1220; 1H NMR (300 MHz, CDCl3) δH: 0.88 (s, 3H), 1.04-1.20 (m, 7H), 1.30-1.44 (m, 7H), 1.48 (m, 2H), 1.50-1.53 (m, 3H), 1.56 (t, 2H, J = 6.96 Hz), 1.60-1.64 (m, 3H), 1.78-1.90 (m, 2H), 3.18-3.22 (m, 2H), 3.50 (t, 2H, J = 13.62 Hz), 3.54 (t, 2H, J = 13.62 Hz), 3.90-4.20 ppm (m, 2H), 4.70 (m, 1H), 6.82 (m, 2H), 6.98 (m, 2H), 7.20-7.40 (m, 4H), 7.64 (m, 2H), 7.70-7.90 (m, 2H), 8.20 (broad, 3H), 8.38 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 14.16, 23.00, 23.10, 24.40, 25.40, 26.00, 26.38, 29.18, 29.80, 32.22, 38.70, 38.86, 39.16, 53.66, 57.90, 59.52, 59.74, 116.70, 117.38, 118.80, 121.36, 122.58, 123.40, 126.78, 128.00, 129.00, 129.24, 130.26, 130.96, 131.04, 132.70, 134.28, 134.40, 152.80, 161.00, 162.28, 163.32, 164.10, 172.22, 176.01 ppm. EI-MS m/z: 797.41 (M+ 11). Anal. Calcd. for C48H55N5O6: C, 72.25; H, 6.95; N, 8.78; O, 12.03. Found: C, 72.18; H, 6.90.2-[Hex-1-ynyl-(3-phenyl-allyl)amino]-3,4-dimethyl-5- phenyl-cyclopent-2-enone (10)A solution of cinnamaldehyde (87 µl, 0.69 mmol), 4-aminoantypirine (100 mg, 0.50 mmol), hexyn-1 (56 µl, 0.50 mmol) and cupric chloride anhydrous (100 mg, 0.74 mmol) in 5 ml of methanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (4:1) yielding 60% of product, m.p. 98-100°C; IR (Vmax, cm-1): 2264, 1720 and 1170; 1H NMR (300 MHz, CDCl3) δH: 0.90 (s, 3H), 1.24 (s, 3H), 1.42 (t, 2H, J = 7.13 Hz), 1.50 (t, 2H, J, 6.00 Hz), 1.78 (s, 3H), 2.30 (t, 2H, J = 6.00 Hz), 2.50 (m, 1H), 3.80 (m, 2H), 4.08 (m, 1H), 5.80-6.80 (m, 2H), 7.20 (m, 5H), 7.30-7.40 (m, 5H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 13.58, 15.00, 16.28, 16.88, 21.94, 32.00, 39.58, 62.00, 62.80, 65.20, 89.48, 126.48, 126.66, 127.10, 127.87, 128.55, 128.80, 130.00, 134.28, 135.68, 137.92, 143.70, 145.74, 198.44 ppm. EI-MS m/z: 397.24 (M+ 10). Anal. Calcd. for C28H31NO: C, 84.59; H, 7.86; N, 3.52; O, 4.02. Found: C, 84.50; H, 7.80.(E)-5-((2-aminoethyl)imino)-N-cinnamyl-N-(hex-1-yn-1-yl)-2,3-dimethyl-4-phenylcyclopent-1-en-1-amine (11)A solution of 10 (200 mg, 0.50 mmol), ethylenediamine (80 µl, 0.74 mmol), and boric acid (40 mg, 0.60 mmol) in 5 ml of methanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (4:1) yielding 54% of product, m.p. 76-78°C; IR (Vmax, cm-1):3382, 3320 and 2260; 1H NMR (300 MHz, CDCl3) δH: 0.90 (s, 3H), 1.14 (s, 3H), 1.46-1.50 (m, 4H), 1.70 (s, 3H), 2.36 (m,2H), 3.10 (t, 2H, J = 6.44 Hz), 3.48 (m, 1H), 3.56 (t, 2H, J = 6.44 Hz), 3.60 (m, 1H), 3.84 (m, 2H), 4.30 (broad, 2H), 5.90-6.80 (m, 2H), 7.00-7.14 (m, 5H), 7.20-7.40 (m, 5H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 13.59, 16.48, 16.60, 16.88, 22.00, 32.00, 40.80, 49.62, 50.44, 54.00, 62.28, 62.50, 89.84, 121.44, 126.18, 126.48, 126.64, 127.80, 128.58, 128.92, 128.85, 130.00, 136.02, 136.54, 139.08, 140.95, 149.56 ppm. EI-MS m/z: 439.29 (M+ 10). Anal. Calcd. for C30H37N3: C, 81.96; H, 8.48; N, 9.56. Found: C, 81.90; H, 8.40.2-Chloro-N-(2-{2-[hex-1-ynyl-(3-phenyl-allyl)-amino]-3,4-dimethyl-5-phenyl-cyclopent-2-enylideneamino}-ethyl-acetamide (12)A solution of 11 (200 mg, 0.45 mmol), triethylamine (100 μl, 1.50 mmol) and chloroacetyl chloride (128 μl, 1.60 mmol) in 5 ml of methanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (4:1) yielding 77% of product, m.p. 115-117°C; IR (Vmax, cm-1): 3378, 2264 and 1680; 1H NMR (300 MHz, CDCl3) δH: 0.90 (s, 3H), 1.14 (s, 3H), 1.44-1.50 (m, 4H), 1.70 (s, 3H), 2.32 (m, 2H), 3.50 (m, 1H), 3.60 (t, 2H, J =6.54 Hz), 3.62 (m, 1H), 3.66 (t, 2H, J =6.54 Hz), 3.90 (m, 2H), 4.00 (m, 2H), 6.00-6.80 (m, 2H), 7.00 (broad, 1H), 7.04-7.15 (m, 5H), 7.20-7.40 (m, 5H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 13.60, 16.46, 16.62, 16.94, 21.98, 32.00, 34.80, 42.47, 49.62, 50.38, 54.32, 62.22, 62.50, 89.70, 121.50, 126.20, 126.52, 126.64, 127.90, 128.63, 128.82, 130.00, 135.88, 136.54, 139.04, 140.82, 149.56, 162.70 ppm. EI-MS m/z: 515.27 (M+ 11). Anal. Calcd. for C32H38ClN3O: C, 74.47; H, 7.42; Cl, 6.87; N, 8.14; O, 3.10. Found: C, 74.40; H, 7.36.3-(2-Hydroxy-naphtalen-yl)-oxirane-2-carboxyl acid (2-{2-[hex-1-ynyl-(3-phenyl-allyl)-amino]-3,4-dimethyl-5-phenyl-cyclopent-2-enylideneamino}-ethyl)-amide (13)A solution of 12 (200 mg, 0.39 mmol), 2-hydroxy-1- naphthaldehyde (68 mg, 0.40 mmol), and sodium hydroxide (20 mg, 0.50 mmol) in 5 ml of ethanol was stirring for 72 h at room temperature. The reaction mixture was evaporated to a smaller volume. After the mixture was diluted with water and extracted with chloroform. The organic phase was evaporated to dryness under reduced pressure, the residue was purified by crystallization from methanol:water (4:1) yielding 65% of product, m.p. 104-106°C; IR (Vmax, cm-1): 3380, 2262, 1684 and 690; 1H NMR (300 MHz, CDCl3) δH: 0.90 (s, 3H), 1.12 (s, 3H), 1.46-1.50 (m, 4H), 1.70 (m, 3H), 2.34 (m, 2H), 3.50 (m,1H), 3.54 (t, 2H, J = 6.54 Hz), 3.60 (m, 1H), 3.66 (t, 2H, J = 6.54 Hz), 3.90 (m, 2H), 3.98-4.30 (m, 2H), 6.00-6.80 (m, 2H), 7.00-7.20 (m, 7H), 7.26-7.34 (m, 2H), 7.36-7.40 (m, 3H), 7.44-7.70 (m, 3H), 7.80 (broad, 2H), 7.90 (m, 1H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 13.60, 16.48, 16.66, 16.90, 22.00, 32.00, 35.51, 49.62, 50.38, 53.72, 54.28, 59.55, 62.20, 62.62, 89.70, 118.80, 121.36, 121.50, 122.62, 123.40, 126.26, 126.48, 126.58, 126.80, 127.90, 128.00, 128.56, 128.85, 129.18, 130.00, 130.42, 134.40, 135.90, 136.52, 139.08, 140.92, 149.60, 152.82, 172.12 ppm. EI-MS m/z: 651.34 (M+ 10). Anal. Calcd. for C43H45N3O3: C, 79.23; H, 6.96; N, 6.45; O, 7.36. Found: C, 79.18; H, 6.90.
3. Results and Discussion
- In this study several straightforward routes are reported for synthesis of two new epoxide-derivatives using some strategies; the first stage (Figure 1 and 2) was achieved by the synthesis of an ether group involved in the chemical structure of compound 4-[4-(3-Butyl-1-cyclohexyl-4-cyclohe-xylimino-azetidin-2-ylidenecarbamoyl)-phenoxy]-benzoic acid (3). It is noteworthy that there are many procedures for preparation of several ether derivatives; however, despite its broad scope, they have some drawbacks; For example, several reagents used are hazardous and expensive such as Iodophenol [9], 1,4-diazabicyclo[2.2.2]octane [10], 2,2,6,6-tetramethyl-heptane-3,5-dione [11] aryltrifluorobora te salts [12]. Another data indicate that formation of ether groups via displacement of nitro groups with hydroxyl groups using a dipolar aprotic solvent; In general, dipolar solvents are used to attain high yield of ether groups [7]. Therefore, in this study, the compound 3 was synthetized by the reaction of 1 with 4-hydroxybenzoic acid in presence of dimetyhylsulfoxide at mild conditions. The 1H NMR spectrum of 3 shows signals at 0.86 ppm for methyl group; at 1.08-1.44, 1.50-1.54, 1.62-1.66 and 3.17-3.20 ppm for cyclohexane ring; at 1.44, 1.58 and 1.78-1.90 ppm for me thylene groups of arm bound to cyclobutane ring; at 4.80 ppm for proton of cyclobutane ring; at 6.94-8.44 for phenyl groups; at 10.80 ppm for carboxyl group. The 13C NMR spectrum of 3 contains peaks at 14.16 for methyl group; at 23.00, 24.40-26.28, 32.20 and 57.88-59.68 ppm for hexane ring; at 23.08 and 29.10-29.69 ppm for methylene groups of arm bound to cyclobutane ring; at 38.70, 134.46 and 164.09 ppm for carbons of cyclobutane ring; at 116.70, 133.84, 152.10 and 165.08 ppm for phenyl groups; at 168.40 ppm for carboxyl group; at 175.95 ppm for amide group. Finally, the presence of compound 3 was confirmed with the mass spectrum which showed a molecular ion at m/z 543.30.
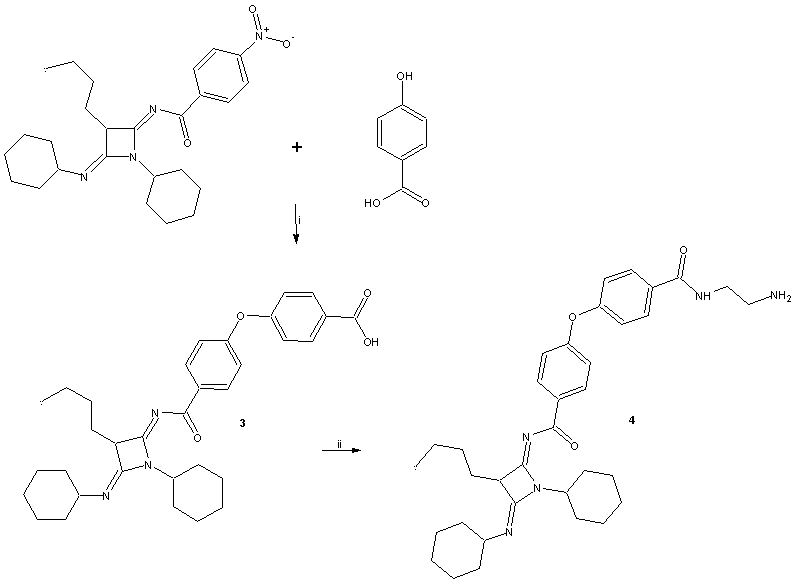 | Figure 1. Synthesis of a phenoxy-carboxamide derivative (4). Reaction of a nitrobenzamide analog (1) with 4-hydroxy benzoic acid (2) to form the cyclohexylimino-azetidin-benzoic acid derivative (3). Following, 3 was reacted with ethylenediamine (ii) in presence of boric acid to form 4. i = K2CO3/rt |
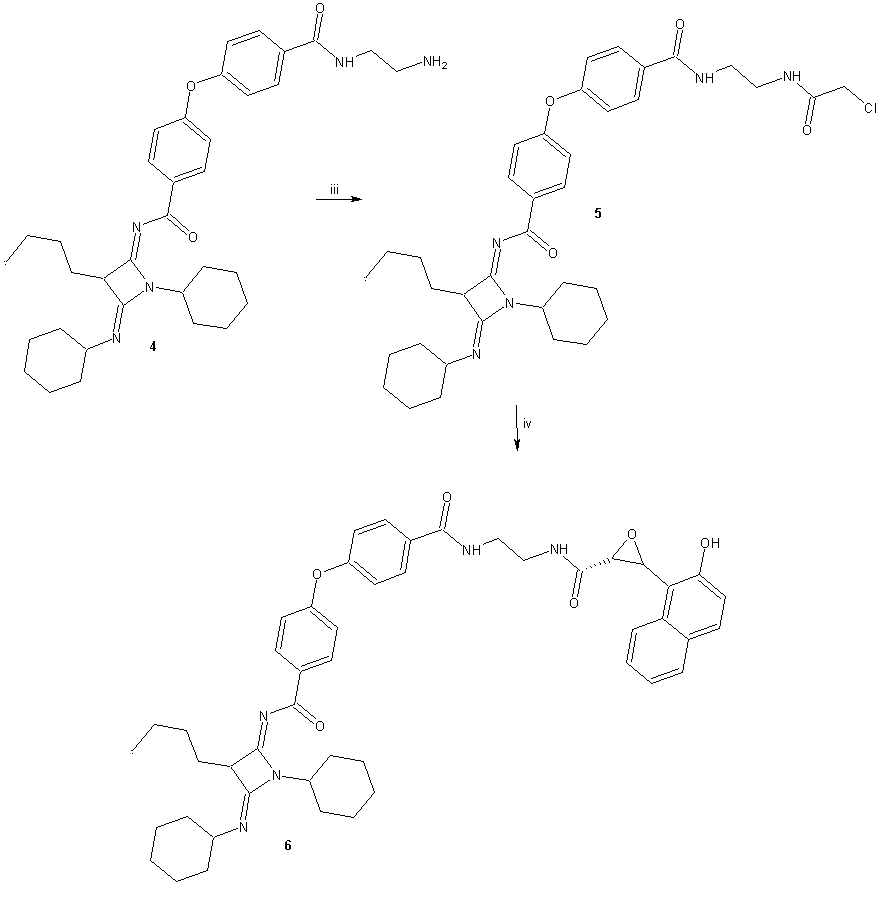 | Figure 2. Synthesis of an epoxide-benzamide derivative (6). Reaction of compound 4 with chloroacetyl chloride in presence of triethylamine (iii) to form a chloroamide derivative (5). After, 5 was reacted with 2-hydroxy- 1-naphthaldehyde (iv) in basic medium to form 6 |
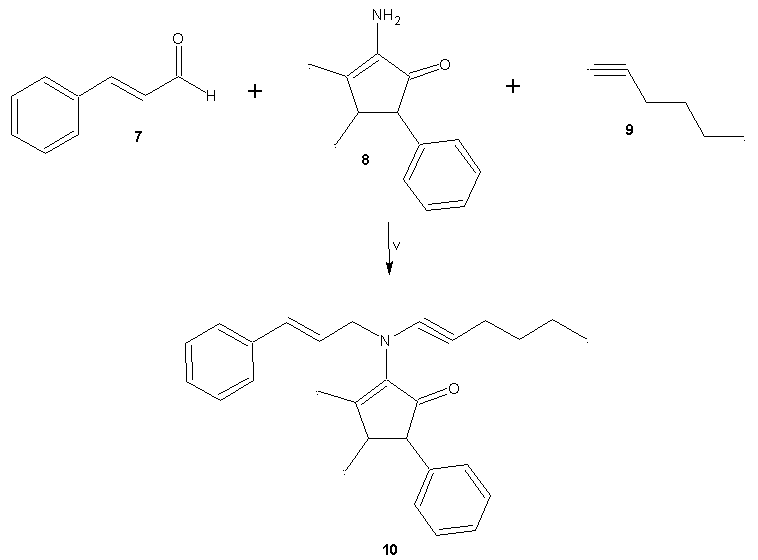 | Figure 3. Synthesis of an enone derivative (10). Reaction of cinnamaldehyde (7), 4-aminoantipyrine (8) and alkyne-1 (9) to form 10. v = cupric chloride anh./MeOH/rt |
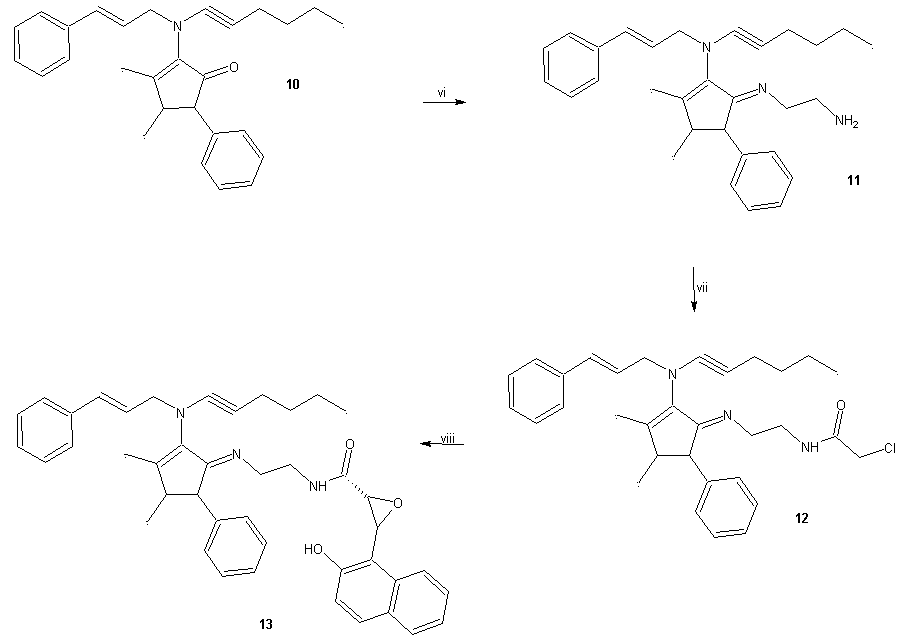 | Figure 4. Synthesis of an epoxide-amide derivative (13). Reaction of 10 with ethylenediamine in presence of boric acid (vi) to form an phenylcyclopentamine derivative (11). After, 11 was reacted with chloroacetyl chloride using treiethylamine as catalyst (vii) to form a chloroacetamide analog (12). Following, 12 was reacted with 2-hydroxy-1-naphthaldehyde (vii) in basic medium for preparation of 13 |
4. Conclusions
- In this study was reported the synthesis of two new epoxide derivatives. The proposed method offers some advantages such as simple procedure and ease of workup.