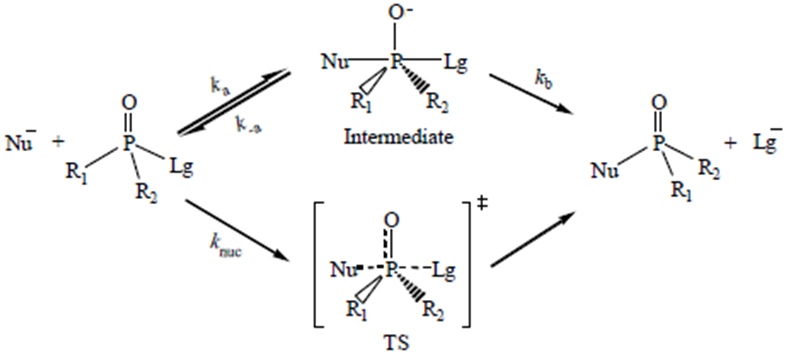
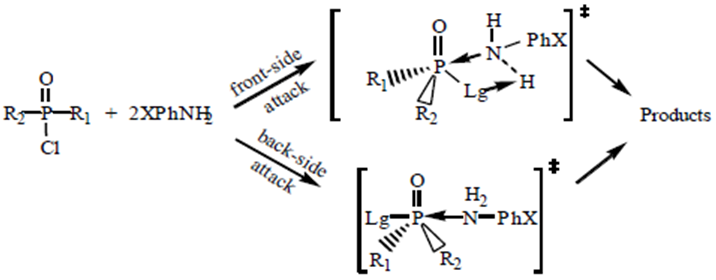
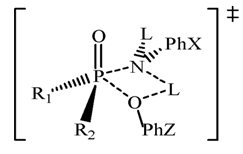
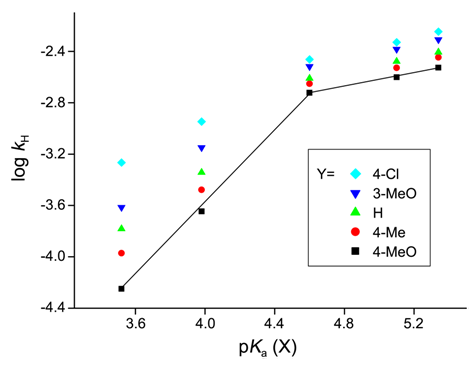
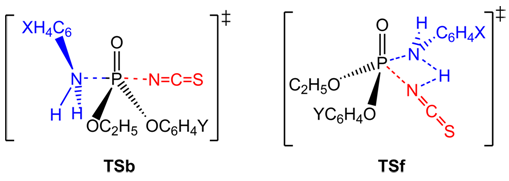
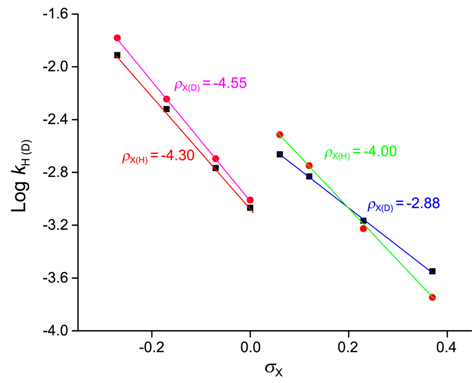
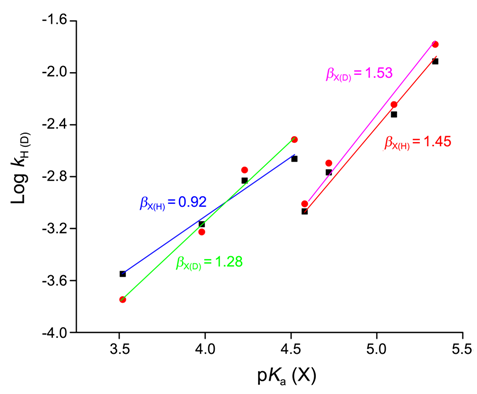
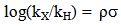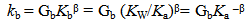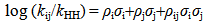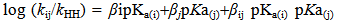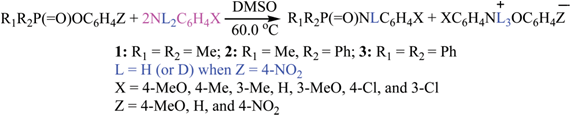| [1] | March, J. “Advanced Organic Chemistry”, 4th edition, Wiley interscience, New York, 1992. |
| [2] | [a] Quin, L.D. “A guide to Organophosphorous Chemistry”, Wiley-Interscience, New York,2000, p 2. [b] Hengge, A. C. 2005, “Mechanistic studies on enzyme-catalyzed phosphoryl transfer” Adv. Phys. Org. Chem., 40, 49-108. |
| [3] | [a] Harger, M.J.P., “Nucleophilic substitution reactions of benzyl- and diphenylmethyl-phosphonamidic chlorides with amines: competition between the usual SN2(P) mechanism and elimination–addition with an alkylideneoxophosphorane (phosphene) intermediate”, J.Chem. Soc., perkin Trans. 2, 2001, 41. [b] Hoque, M.E.U.; Dey, S.; Kim, C.K. and Lee, H.W. “Kinetics and Mechanism of the Pyridinolysis of Aryl Phenyl Chlorothiophosphates in Acetonitrile”, Bull. Korean Chem. Soc. 2011,32,1138. [c] Domingos, J. B.; Longhinotti, E.; Buntor, C. A. and Nome, F.J. Org. Chem., 2003,68,305. [d] Harger, M. J. P., “Methylenethioxophosphorane (thiophosphene) Intermediates in the Reactions of Diphenylmethyl phosponamidothioic Chlorides with amines. Differences between the Elimination – Addition Mechanisms of Nucleophilic Substitution for P=S and P=O Substrates”, J. Chem. Soc. Perkin Trans. 2: 2002, 489. [e] Hengge, A. C., “Isotope effects in the Study of Phosphoryl and Sulfuryl Transfer Reactions”, Acc. Chem. Res., 2002, |
| [4] | Williams, A., “Free Energy Relationship in Organic and Bio-Organic Chemistry”, (UK: RSC Press), 2003. |
| [5] | [a] Lee, I., “Characterization of Transition States for Reaction in Solution by Cross Interaction Constants.”1990, Chem. Soc. Rev.,19, 317. [b] Lee, I., 1992. “Cross-Interaction Constants and Transition State Structures”, Adv. in Phys. Org. Chem., 27, 57. [c] Lee, I., “Secondary Kinetic Isotope Effects Involving Deuterated Nucleophiles” Chem. Soc. Rev.1995, 24, 223. [d] Lee, I. and Lee, H. W., “Cross-Interaction Constants, a Mechanistic Tool.” Coll.Czech.Chem.Commun. 1999, 64, 1529. [e] I. Lee, and Lee, H. W., “Cross Interaction Constant and Intrinsic Barrier”, Bull. of Korean Chem. Soc. 2001, 22, 732. |
| [6] | (a) Olaf, W.; Houk, K. N.; Black, K. A. and Thomas, B., J. Am.Chem. Soc., 1995, 117, 8594. (b) Carrol, F. A., “Perspectives on Structure and Mechanism in Organic Chemistry (ITP)”, 1998. (c) Isaacs, N. S., “Physical Organic Chemistry(Longman: Harlow, 2nd ed )”,1995.(d) Paneath, P. and O’Leary, M. H., J. Am.Chem.Soc.,1991,113,1691. |
| [7] | [a] Adhikary, K. K.; Lumbiny, B. J.; Dey, S. and Lee, H. W.,, “ Transition State Variation in the Anilinolysis of O-Aryl Phenyl Phosphonochloridothioates in Acetonitrile”. Bull. of Korean Chem. Soc.,2011, 32, 2628. [b] Lumbiny, B. J. and Lee, H. W., “Anilinolysis of S-Aryl Phenyl Phosphonochloridothioates in Acetonitrile,” Bull. of Korean Chem.Soc.,2008, 29,2065. [c] Hoque, M. E. U.; Dey, S.; Guha, A. K.; Kim, C. K.; Lee, B. S. and Lee, H. W., “Kinetics and Mechanism of the Aminolysis of the Aryl Phenyl Chlorophosphates with Anilines”, J. Org. Chem.,2007, 72,5493. |
| [8] | [a] Zalatan, J.; Herschlag, D.,“ Alkaline Phosphatase Mono- and Diesterase Reactions: Comparative Transition State Analysis”, J. Am. Chem. Soc., 2006,128, 1293-1303. [b] O’Brien, P. J.; Herschlag, D., “Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase,” Biochemistry, 2001, 40, 5691-5699. [c] Catrina, I. E., Hennge, A. C., “The effect of divalent metal ions on the rate and transition-state structure of phosphoryl-transfer reactions”, J. Am. Chem. Soc., 1987,109, 4665-4674. |
| [9] | Hansch, C.; Leo, A. and Taft, R. W. “Survey of Hammett Substituent Constants and Resonance and Field Parameters”, Chem. Rev.,1991. 91,165-195. |
| [10] | Zuman, P. and Patel, R. C., “Techniques In Organic Reactions Kinetics”,(John Wiley and Sons, USA), 1984. |
| [11] | Albert, A. and Serjeant, E. P., “The Determination of Ionization”, (Chapman and Hall, New York, 3rd ed.),1984. |
| [12] | [a] Lee, I.; Kang, H. K. and Lee, H. W., J. Am. Chem. Soc.,1987, 109, 7472. [b] Lee, I.; and Lee, H.W., “Cross-interaction constants as ameasure of the transition-state structure. 14. Nucleophilic substitution reactions of 1-phenyl-2-propylbenzenesulfonates with anilines in methanol”, J. Org. Chem., 1991, 56, 4682. [c] I. Lee, “Bimolecular nucleophilic substitution at a resonance-stabilized carbenium ion: Elevated value of cross-interaction constant, imbalanced transition state and the non-interactive phenomenon”, J. Phy. Org. Chem., 1994,7, 448. [d] Guha, A.K.; Lee, H.W. and Lee, I., 1999, “Kinetics and mechanism of the aminolysis of phenyl substituted phenyl chlorophosphates with anilines in acetonitrile”, I. J. Chem. Soc., Perkin Trans. 2, 1999, 765. |
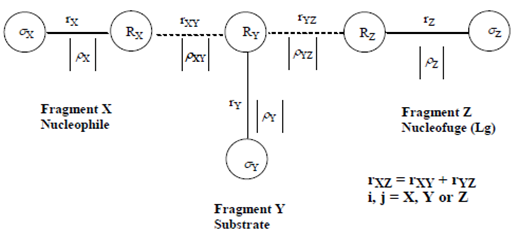
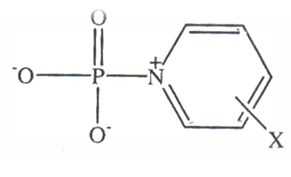
| [13] | Bourne, N. and Williams, A. “Evidence for a single transition state in the transfer of the phosphoryl group (-PO32-) to nitrogen nucleophiles from pyridinio-N-phosphonates” J. Am. Chem. Soc., 1984, 106, 7591. |
| [14] | Bourne, N.; Chrystiuk, F.; Davis, A. M. and Williams, A. “A single transition state in the reaction of aryl diphenylphosphinate esters with phenolate ions in aqueous solution”, J. Am. Chem. Soc., 1988, 110,1890. |
| [15] | Guha, A.K.; Lee, H.W. and Lee, I., “Pyridinolysis of Phenyl- Substituted Phenyl Chlorophosphate in Acetonitrile”, 2000, J. Org. Chem., 65, 12-15. |
| [16] | Dewar, M. J. S. “The Molecular orbital Theory of Organic Chemistry”, McGraw-Hill:New York, 1969,p 358. |
| [17] | Lee, H. W. and Guha, A. K., Lee, I., “Kinetics and mechanism of the reaction of para-chlorophenyl aryl chlorophosphates with anilines in acetonitrile”, Int. J. Chem. Kinet, 2002, 34,632. |
| [18] | Lee, I.;, Kim, C. K.; Li, H. G.; Sohn, C. K.;C. K. Kim, H. W. Lee and B. S. Lee, “Acyl-Transfer Mechanisms Involving Various Acyl Functional Groups:  with X = C, S, P and Y = O, S”, J. Am.Chem.Soc., 2000, 122, 11162. with X = C, S, P and Y = O, S”, J. Am.Chem.Soc., 2000, 122, 11162. |
| [19] | Um, I.H.; Shin, Y. H; Han, J. Y. and Mishima, M. “Aminolysis of Y- Substituted Phenyl Diphenyl Phosphinates and Benzoates: Effect of Modification of Electrophilic center from C=O to P=O” J. Org. Chem, 2006, 71, 7715-7720. |
| [20] | Hoque, M.E.U.; Dey, N. K.;Kim, C. K., Lee, B. S. and Lee, H. W. “Kinetics and mechanism of the aminolysis of aryl ethyl chloro and chlorothio phosphates with anilines”, Org. Biomol. Chem., 2007,5, 3944-3950. |
| [21] | Um, G. H.; Han, J. Y. and Shin, Y. H. “Aminolysis of X-Substituted Phenyl Diphenylphosphinates: Effect of Amine Nature on Reactivity and Transition-State Structure”, J. Org. Chem, 2009, 74, 3073-3078. |
| [22] | [a] Dey, N.K.; Hoque, M. E. U. ; Kim, C. K.; Lee, B. S. and Lee, H. W. “ Kinetics and mechanism of the anilinolysis of dimethyl and diethyl chloro(thino) phosphates”, J.Phy.Org.Chem., 2008,21, 544-548. [b] Hoque, M. E. U. and Lee, H. W., “Kinetics and Mechanism of the Anilinolysis of Dipropyl Chlorophosphate in Acetonitrile, Bull. Korean. Chem. Soc., 2012,33 ( 6) 1879. [c] Hoque, M. E. U. andLee, H. W., “Kinetics and Mechanism of the Anilinolysis of Dibutyl Chlorophosphate in Acetonitrile, Bull. Korean. Chem. Soc., 2012,33( 2) 663. [d] Hoque, M. E. U. and Lee, H. W., “Kinetics and Mechanism of the Anilinolysis of Diisopropyl Chlorophosphate in Acetonitrile, Bull. Korean. Chem. Soc., 2011, 32(9), 3245. |
| [23] | Taft, R. W. “Steric Effects in Organic Chemistry”, Newman, M.S., Wiely, New York, Chapter 3.1956. |
| [24] | Hoque, M. E. U. and Lee, H. W., “Pyridinolysis of Dibutyl Chlorophosphate in Acetonitrile”, 2012,33( 3), 1055. |
| [25] | [a] Dey, N.K.; Hoque, M. E. U.; Kim, C. K.; Lee, B. S. and Lee, H. W., “ Kinetics and mechanism of the aminolysis of dimethyl and methyl phenyl phosphinic chlorides with anilines”, J.Phy.Org. Chem., 2009, 22, 425-430.[b] Dey, N. K. and Lee, H. W., “Anilinolysis of diethyl pphosphinic chloride in Acetonitrile”, Bull. Korean. Chem. Soc., 2010,31(5), 1403. [c] Hoque, M. E. U. and Lee, H. W. “Kinetics and Mechanism of the Aminolysis of Diphenyl Phosphinic Chloride with Anilines”, Bull. Korean Chem. Soc.,2007,Vol. 28, No. 6, 936-940. [d] Hoque, M. E. U. and Lee, H. W., “Kinetics and Mechanism of the Anilinolysis of Dicyclohexyl phosphinic Chloride in Acetonitrile”, Bull. Korean Chem. Soc.,2011, Vol. 32, 6, 1997. |
| [26] | Hong, H.J.; Lee, J.; Bae, A. R. and Um, I. H., “Kinetics and Reaction Mechanism for Alkaline Hydrolysis of Y-Substituted-Phenyl Diphenylphosphinates”, Bull. Korean Chem. Soc., 2013, Vol. 32, 6 , 2001-2005. |
| [27] | Barai, H. R. and Lee, H. W., “Kinetics and Mechanism of the Aminolyses of Bis(2-oxo 3-oxazolidinyl) Phosphinic Chloride in Acetonitrile”, Bull. Korean Chem. Soc.,2013, Vol. 34, 11. |
| [28] | Cook, R. D.; Daouk, W. A.; Hajj, A. N.; Kabbani, A.; Kurku, A.; Samaha, M.; Shayban, F. and Tanielian, O. V.,Can. J. Chem., 1986, 64, 213- 219. |
| [29] | [a] Cook, R. D.; Rahhal-Arabi, L., “The kinetics of the alkaline hydrolysis of aryl diphenylphosphinothioates; the significance for the mechanism of displacement at phosphorus”, Tetrahedron Lett.,1985,26, 3147-3150. [b] Haake, P.; McCoy, D. R.; Okamura, W.; Alpha, S. R.; Wong, S. W.; Tyssee, D. A.; McNeal, J. P. and Cook, R. D., “Phosphinic acids and derivatives. 4. Linear free energy relationships in displacement at phosphinyl phosphorus” Tetrahedron Lett. 1968, 5243-5246. |
| [30] | [a] Williams, A. and Naylor, J., , “Hydrolysis of phosphinic esters: general-base catalysis by imidazole” J. Chem. Soc.1971, 1967-1972. [b] Buncel, E.; Albright, K. G. and Onyido, I., “Nucleophilic displacement on 4-nitrophenyl dimethyl phosphinate by ethoxide ion: alkali metal ion catalysis and mechanism”, 2004,2, 601-610. |
| [31] | Domingos, J. B.; Longhinotti, E.; Bunton C. A. and Nome, F, “Reactions of bis(2,4-dinitrophenyl) phosphate with hydroxylamine”, J. Org. Chem., 2003, 68, 7051-7058. |
| [32] | [a] Barai, H. R. and Lee, H. W., “Kinetics and Mechanism of the Anilinolysis of Bis(aryl) Chlorophosphates in Acetonitrile”, Bull. Korean Chem. Soc., 2011, 32(6), 1939. [b] Barai, H. R.; Lee, H. W., “Kinetics and Mechanism of the Anilinolysis of Bis(2,6-dimethylphenyl) Chlorophosphate in Dimethyl Sulfoxide”, Bull. Korean Chem. Soc., 2011, 32(10), 3783. |
| [33] | [a] Dey, N. K.; Kim, C. K. and H. W. Lee, “Kinetics and mechanism of the anilinolyses of aryl dimethyl, methyl phenyl and diphenyl phosphinates”, Org. Biomol. Chem., 2011,9, 717. [b] Douglas, K. T. and Williams, A., “Nucleophilic versus general base catalysis in phosphyl (PV) group transfer: application to α-chymotrypsin action”, J. Chem. Soc, Perkin Trans 2., 1976,515. |
| [34] | Barai, H. R.; Adhikary, K. K. and Lee, H.W., “Kinetics and Mechanism of the Anilinolysis of Aryl Ethyl Isothiocyanophosphates in Acetonitrile”, Bull. Korean Chem. Soc., 2013, 34( 6), 1829. |
| [35] | See Reference 27. |
| [36] | Lee, H. W.; Guha, A. K.; Kon C. K.and Lee, I., “Transition-State Variation in the Nucleophilic Substitution Reactions of Aryl Bis(4-methoxyphenyl) Phosphates with Pyridines in Acetonitrile”, J. Org. Chem., 2002,67, 2215. |
| [37] | [a] Rowel, R.; Gorenstein, D. G., “Multiple structure-reactivity correlations in the hydrolysis of epimeric 2-aryloxy-2-oxy-dioxaphosphorinanes. Stereoelectronic effects”, J. Am., Chem. Soc., 1981, 103, 5894. [b] Ba-saif, S. A.; Waring, M. A. and Williams, A. “Single transition state in the transfer of a neutral phos;phoryl group between phenoxide ion nucleophiles in aqueous solution”, J. Am. Chem. Soc., 1990,112, 8115. |
| [38] | Adhikary, K. K.; Lee, H. W. and Lee, I., “Kinetics and Mechanism of the Pyridinolysis of Aryl Phenyl Isothiocyanophosphate in Acetonitrile”, Bull. Korean. Chem. Soc.,2003,24, 1135. |
| [39] | Barai, H. R.; Adhikary, K. K. and Lee, H. W., “Anilinolysis of diethyl isothiocyanophosphate in acetonitrile.” Bull. Korean. Chem. Soc.,2012, 33, 3, 1089. |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML